CASOS CLÍNICOS
Síndrome de Gorlin y afectación ocular
María del Valle Acuña, Mariela Ogusuku, Walter Briggiler
Hospital San Martín, Paraná (Entre Ríos), Argentina.
Recibido: 10 de diciembre de 2024.
Aprobado: 5 de mayo de 2025.
Autor corresponsal
Dra. María del Valle Acuña.
Servicio de Oftalmología del Hospital San Martín
(3100) Paraná, Entre Ríos
Argentina.
+54 (0343) 423-4545
valle_a92@hotmail.com
Oftalmol Clin Exp (ISSNe 1851-2658)
2025; 18(2): e236-e242.
https://doi.org/10.70313/2718.7446.v18.n2.424
Resumen
Objetivos: Describir las características clínicas y oculares del síndrome de Gorlin a propósito de un caso.
Caso clínico: Se recibió interconsulta por paciente masculino de 58 años con antecedente de melanoma facial. Presentaba miasis frontotemporal izquierda sobre lesión hiperpigmentada y afectación palpebral, además de malformación en piezas dentarias y múltiples lesiones hiperpigmentadas nodulares faciales. La visión estaba severamente comprometida. Tras realizar interconsulta con los servicios de cirugía de cabeza y cuello, oncología y odontología se arribó al diagnóstico presuntivo de síndrome de Gorlin en base a los signos multisistémicos observados. El paciente abandonó su atención regresando a los tres meses con empeoramiento del cuadro. En una tomografía computada se observó una masa ocupante en el vértice de la órbita izquierda. Se decidió su internación, por contexto clínico-social del paciente. Se realizó toilette quirúrgica, toma de biopsias y se evaluaron futuras pautas terapéuticas, incluyendo como opciones el uso de vismodegib, pero no se pudieron realizar ya que el paciente firmó su alta voluntaria y no ha vuelto a la institución.
Conclusión: En el caso presentado se lograron identificar mediante interconsultas entre diferentes especialidades las características principales del síndrome de Gorlin: carcinomas basocelulares en diversas regiones, tumores queratoquísticos mandibulares y anomalías esqueléticas junto con las manifestaciones oculares. Lamentablemente por condiciones sociales no se logró realizar el seguimiento del caso.
Palabras clave: síndrome de Gorlin.
Gorlin’s syndrome and ocular manifestation
Abstract
Objective: To describe the clinical and ocular features of Gorlin’s syndrome, based on a case report.
Case report: A 58-year-old male patient with a history of facial melanoma, who had left frontotemporal myiasis on hyperpigmented lesion and palpebral involvement, in addition to malformation of teeth and multiple facial nodular hyperpigmented lesions was consulted. Vision was severely compromised. After consultation with the head and neck surgery, oncology and dentistry services, a presumptive diagnosis of Gorlin’s syndrome was made, based on the multisystemic signs observed. The patient left the clinic and returned three months later with worsening of the fourth episode. A CT scan showed a mass occupying the vertex of the left orbit. It was decided to hospitalize him, due to the clinical and social context of the patient. Surgical toilette was performed, biopsies were taken and future therapeutic guidelines were evaluated, including the use of Vismodegib as an option, but they could not be performed since the patient signed his voluntary discharge and has not returned to the institution.
Conclusion: In the case presented, the main characteristics of Gorlin’s syndrome were identified by means of interconsultations between different specialties: basal cell carcinomas in different regions, mandibular keratocystic tumors and skeletal anomalies, together with ocular manifestations. Unfortunately, due to social conditions it was not possible to follow up the case.
Keywords: Gorlin’s syndrome.
Síndrome de Gorlin e envolvimento ocular
Resumo
Objetivos: Descrever as características clínicas e oculares da síndrome de Gorlin com base em um caso.
Caso clínico: Recebeu-se interconsulta médica de paciente masculino, 58 anos, com histórico de melanoma facial. Apresentava miíase frontotemporal esquerda sobre lesão hiperpigmentada e comprometimento palpebral, além de malformações dentárias e múltiplas lesões faciais hiperpigmentadas nodulares. Sua visão estava gravemente comprometida. Após consulta com os departamentos de cirurgia de cabeça e pescoço, oncologia e odontologia, foi feito o diagnóstico presuntivo de síndrome de Gorlin com base nos sintomas multissistêmicos observados. O paciente abandonou o tratamento e retornou três meses depois com piora dos sintomas. Uma tomografia computadorizada revelou uma massa ocupando o ápice da órbita esquerda. A decisão de interná-lo foi tomada com base no contexto clínico e social do paciente. Foi realizado toilette cirúrgico, biópsias foram realizadas e futuros regimes terapêuticos foram avaliados, incluindo o uso de vismodegib, mas estes não puderam ser implementados porque o paciente assinou um termo de alta voluntária e não retornou à instituição.
Conclusão: No caso apresentado, as principais características da síndrome de Gorlin foram identificadas por meio de consultas entre diferentes especialistas: carcinomas basocelulares em diversas regiões, tumores ceratocísticos da mandíbula e anormalidades esqueléticas, além de manifestações oculares. Infelizmente, devido a circunstâncias sociais, o acompanhamento do caso não foi possível.
Palavras-chave: síndrome de Gorlin.
Introducción
En 1960, Gorlin y colaboradores publicaron lo que potencialmente estimaban que se trataba de un nuevo síndrome en un paciente donde identificaron disostosis craneofacial, conducto arterial persistente, hipertricosis, hipoplasia de labios mayores, anomalías dentales y oculares1. En las siguientes décadas siguieron apareciendo publicaciones donde se identificaban varias características de la apreciación inicial publicada en 1960, pero donde también se agregaban variantes hasta que en 1985 se sumó la afectación cutánea y su relación con el carcinoma basocelular múltiple, además de los tumores queratoquísticos ondontogenéticos2. Finalmente a este síndrome se lo renombró como “síndrome de Gorlin-Goltz”, que también es conocido como “síndrome de carcinoma basocelular nevoide”, donde la más reciente revisión de este tema y la afectación ocular expresa que es una condición genética rara que predispone a las personas a varias anomalías del desarrollo y a tumores3. A nivel ocular se ha confirmado la presencia de hipertelorismo, cataratas congénitas y nistagmus, destacando la alta frecuencia de estrabismo, membranas epirretinales y mielinización del nervio óptico3.
Si bien han pasado casi seis décadas y media desde que se iniciaron los primeros informes de este síndrome, quedan muchos interrogantes y las evidencias publicadas son pocas. Se trata de una enfermedad rara con una prevalencia estimada que oscila entre 1/30.827 a 1/256.0001-5, donde se afectan por igual hombres y mujeres4. Es una enfermedad de base genética autosómica dominante que tiene alta variación fenotípica intrafamiliar causada por mutaciones de la línea germinal del gen PTCH15-6. Aproximadamente entre el 50% y el 85% de los casos se origina por variantes patogénicas del gen supresor de tumores PTCH16, localizado en cromosoma 9 (9q22.3) pero entre el 15% y el 27% de los casos aún se desconoce la causa6-7.
Por tal motivo, el objetivo de nuestro trabajo ha sido realizar la descripción de los hallazgos encontrados en un paciente con diagnóstico presuntivo de síndrome de Gorlin y compartir las dificultades que nuestro grupo tuvo que enfrentar en el manejo del caso clínico.
Caso clínico
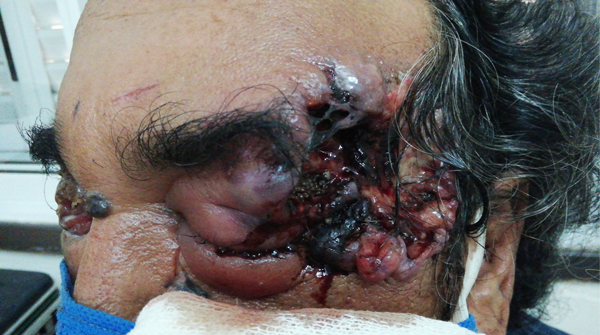
Se recibió interconsulta en el servicio oftalmológico del Hospital San Martín de la ciudad de Paraná (Entre Ríos, Argentina) por paciente masculino de 58 años con antecedente de melanoma facial (diagnosticado en 1998), que concurrió por guardia central del hospital con miasis fronto-temporal izquierda sobre lesión hiperpigmentada que comprometía párpados.
En la valoración inicial se observó la malformación de múltiples piezas dentarias y se constataron múltiples lesiones hiperpigmentadas nodulares faciales de las cuales emergían parásitos larvarios. En el examen ocular se constató que la agudeza visual en ojo derecho (OD) era visión bulto y en ojo izquierdo (OI) no se pudo evaluar por edema bipalpebral a tensión (figs. 1 y 2). Se realizó laboratorio clínico, tomografía computada (TC) de cerebro y órbitas, además de una ecografía ocular de ambos ojos, que no arrojaron anormalidades inicialmente. Se decidió realizar interconsulta con los servicios de cirugía de cabeza y cuello, oncología y de odontología. Luego de debatir el caso, se arribó al diagnóstico presuntivo de síndrome de Gorlin en base a los signos multisistémicos observados.
Figura 1. Lesión dérmica en área temporal izquierda severa con áreas de necrosis tisular en contexto de paciente con miasis y edema bipalpebral.
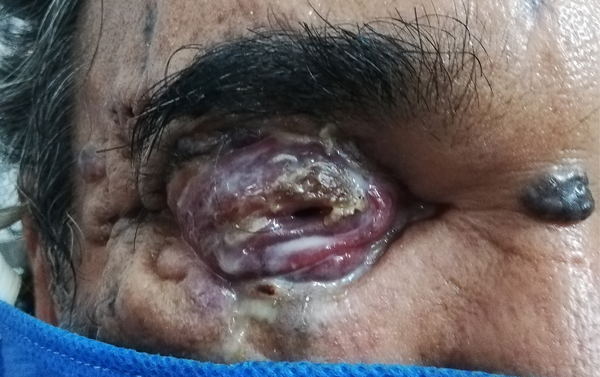
Figura 2. Severa afectación de región periocular derecha con secreción purulenta y múltiples lesiones pigmentarias en piel circundante.
Desafortunadamente el paciente abandonó su atención y regresó a los tres meses con empeoramiento del cuadro general, principalmente a nivel ocular (figs. 3 y 4), más allá de una mejoría parcial de la lesión dérmica temporal izquierda y la resolución del severo edema bipalpebral izquierdo del primer control.
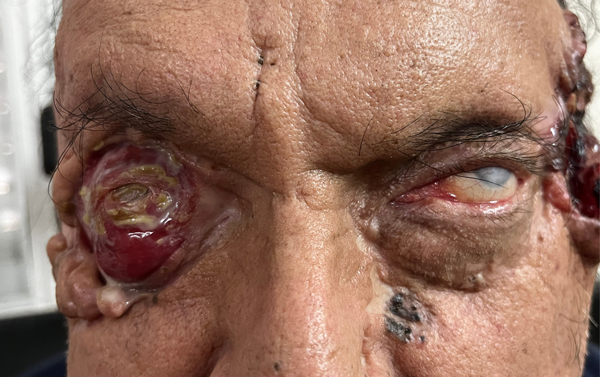
Figura 3. Empeoramiento de lesión ocular derecha; en la misma fotografía se observa que el edema bipalpebral izquierdo logró resolverse luego del tratamiento inicial.
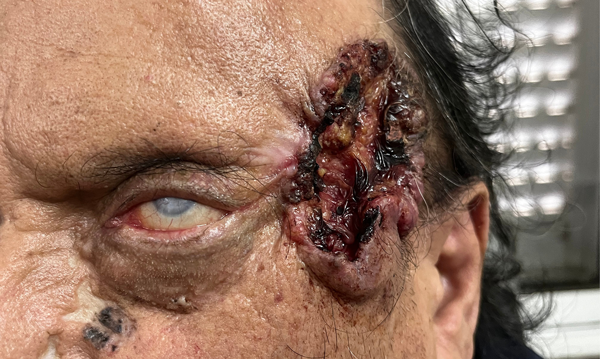
Figura 4. Evolución favorable de lesión dérmica temporal izquierda.
Al repetir la TC se observó una masa ocupante en el vértice de la órbita izquierda. Se decidió su internación debido al contexto clínico-social del paciente. Se realizó toilette quirúrgica, toma de biopsias y se evaluaron futuras pautas terapéuticas, incluyendo otras opciones como la utilización de vismodegib y la intención de buscar la confirmación genética del diagnóstico. Nuevamente se perdió el seguimiento del paciente, quien firmó su alta voluntaria y no volvió a la institución.
Discusión
El presente caso resultó un gran desafío para el equipo hospitalario, tanto para establecer su diagnóstico como para su manejo y contención. En el ámbito hospitalario, el manejo de los pacientes incluye la consideración de los problemas socio-culturales y económicos de la población, pero para los médicos es muy complejo poder dar solución a esos aspectos que rodean a las enfermedades y a los propios pacientes. El caso presentado resume tanto las problemáticas de personas en situación de calle, alteraciones psíquicas y falta de contención social, situaciones para los que la salud pública hace lo que puede, comprendiendo que no siempre es suficiente, cuando además aparecen patologías raras o poco frecuentes.
Moramarco y colaboradores son quienes más han aportado en la literatura científica sobre las manifestaciones oculares del síndrome de Gorlin-Goltz en un estudio que evaluó a once pacientes en quienes se confirmó esta patología mediante test genéticos3. Ellos encontraron en su casuística alteraciones como hipertelorismo, cataratas congénitas, nistagmus, estrabismo, membranas epirretinales y mielinización del nervio óptico, concluyendo que las características oculares —que son frecuentes y distintivas— podrían ayudar en el proceso de diagnóstico de este síndrome, poniendo énfasis en el papel crucial del oftalmólogo en el manejo de estos pacientes. Esto fue en parte lo que nos motivó también a nuestro grupo al desarrollo del presente trabajo, pero en nuestro caso tuvimos limitaciones tanto a la hora de poder realizar la confirmación diagnóstica genética como en el momento del seguimiento del paciente por los aspectos previamente mencionados. En cuanto al tratamiento, en nuestro caso fue en todo momento orientado a resolver las complicaciones, el cuadro de miasis y el manejo de las lesiones dérmicas sin muchas más opciones para realizar debido al grado de afección que tenía el paciente en el momento de la primera consulta.
Se llegó a valorar la posibilidad de utilizar el vismodegib de acuerdo con lo que se recomienda en la revisión sistemática publicada en 2024 por Palmeiro y colaboradores8, algo que, si consideramos la falta de adhesión del paciente a la hora de realizar sus controles, posiblemente también nos hubiera llevado al fracaso. Esto no alcanzamos a realizarlo.
Encontramos un informe de alteración psiquiátrica en relación con el síndrome de Gorlin en un adulto de 34 años, pero no hay mayor información para realmente establecer una asociación, aunque al considerar el caso de nuestro paciente estimamos que también podría haber tenido una alteración que no pudo ser valorada: el potencial desorden mental del caso podría haber sido reactivo, influenciado por su historia de vida o incluso por una afectación intracraneal secundaria. Finalmente, en el contexto de la masa ocupante en el vértice de la órbita y que dentro de este síndrome se incluye la presencia de carcinomas basocelulares múltiples, el futuro y la expectativa de vida de nuestro paciente pareciera ser limitado, más allá de las lesiones oculares.
Conclusión
Al coordinar las interconsultas entre especialidades en el caso presentado se lograron identificar las principales características clínicas del síndrome de Gorlin: carcinomas basocelulares en diversas regiones, tumores queratoquísticos mandibulares y anomalías esqueléticas, junto con las manifestaciones oftalmológicas. Lamentablemente, a pesar de poder realizar un diagnóstico clínico presuntivo, no se ha logrado contener al paciente y continuar con su tratamiento por sus condiciones sociales.
Referencias
1. Gorlin RJ, Chaudhry AP, Moss ML. Craniofacial dysostosis, patent ductus arteriosus, hypertrichosis, hypoplasia of labia majora, dental and eye anomalies: a new syndrome? J Pediatr 1960; 56: 778-785. doi:10.1016/s0022-3476(60)80315-0.
2. MacSweeney JE, Forbes A, Manhire AR, Lees WR. Gorlin's syndrome. J R Soc Med 1985; 78(3): 253-255. doi:10.1177/014107688507800315.
3. Moramarco A, Himmelblau E, Miraglia E et al. Ocular manifestations in Gorlin-Goltz syndrome. Orphanet J Rare Dis 2019; 14(1): 218. doi:10.1186/s13023-019-1190-6.
4. Lo Muzio L, Nocini PF, Savoia A et al. Nevoid basal cell carcinoma syndrome: clinical findings in 37 Italian affected individuals. Clin Genet 1999; 55(1): 34-40. doi:10.1034/j.1399-0004.1999.550106.x.
5. Farndon PA, Del Mastro RG, Evans DG, Kilpatrick MW. Location of gene for Gorlin syndrome. Lancet 1992; 339(8793): 581-582. doi:10.1016/0140-6736(92)90868-4.
6. Bholah Z, Smith MJ, Byers HJ, Miles EK, Evans DG, Newman WG. Intronic splicing mutations in PTCH1 cause Gorlin syndrome. Fam Cancer 2014; 13(3): 477-480. doi:10.1007/s10689-014-9712-9.
7. Evans DG, Oudit D, Smith MJ et al. First evidence of genotype-phenotype correlations in Gorlin syndrome. J Med Genet 2017; 54(8): 530-536. doi:10.1136/jmedgenet-2017-104669.
8. Palmeiro AG, Carvalho M, Gonçalves Castro C, Pimentel B, Catorze G. Vismodegib in Gorlin-Goltz syndrome: a systematic review. Australas J Dermatol 2024; 65(6): e123-e133. doi:10.1111/ajd.14326.
9. Mufaddel A, Alsabousi M, Salih B, Alhassani G, Osman OT. A case of Gorlin-Goltz syndrome presented with psychiatric features. Behav Neurol 2014; 2014: 830874. doi:10.1155/2014/830874.