
CASOS CLÍNICOS
Retinosis pigmentaria sine pigmento unilateral: a propósito de un caso
Eden Belmont Wasserlauf, Dolores Varela Fuentes, Agustín Ignacio Fernández
Servicio de Oftalmología, HIGA San Martín, La Plata, Argentina.
Oftalmol Clin Exp (ISSNe 1851-2658)
2024; 17(3): e433-e439.
DOI:10.70313/2718.7446.v17.n03.353
Resumen
Nuestro objetivo es presentar un caso de retinosis pigmentaria unilateral sine pigmento. Se trata de un paciente de sexo femenino de 41 años de edad que fue evaluada al referir disminución de la agudeza visual de ojo derecho (OD) de 2 años de evolución. Al examen físico, agudeza visual en OD 7/10 mejor corregida y en ojo izquierdo 10/10 sin corrección. A la biomicroscopía, en OD se evidenciaba una catarata subcapsular posterior estrellada y en el fondo de ojos se constató estrechamiento arteriolar, alteración del epitelio pigmentario de la retina y lesiones blanquecinas en región macular que también se encontraban en la periferia retinal. El campo visual computarizado del OD mostró un patrón de contracción, mientras que el electrorretinograma indicó una respuesta subnormal en condiciones escotópicas en el OD. Por otro lado, los estudios realizados en el ojo izquierdo se encontraron dentro de los parámetros de normalidad. En conclusión, la presentación de un caso de retinosis pigmentaria sine pigmento unilateral es extremadamente inusual, lo que nos motivó a estudiar en profundidad este caso.
Palabras clave: retinosis pigmentaria, sine pigmento, unilateral.
Unilateral retinitis pigmentosa sine pigmento: a case report
Abstract
Our objective is to present a case of unilateral retinitis pigmentosa (RP) sine pigmento, a 41-year-old female patient evaluated at our institution with a 2-year evolution of decreased visual acuity in the right eye (RE). On physical examination, visual acuity in right eye 7/10 best corrected and in left eye 10/10 without correction. Biomicroscopy in the right eye showed a posterior stellate subcapsular cataract and fundus examination revealed arteriolar narrowing, alteration of the retinal pigment epithelium and whitish lesions in the macular region, which were also found in the retinal periphery. The computed visual field of the RE showed a contraction pattern, while the electroretinogram indicated a subnormal response under scotopic conditions in the RE. On the other hand, studies performed in the LE were found to be within normal parameters. In conclusion, the presentation of a case of unilateral retinitis pigmentosa sine pigmento is extremely unusual, which motivated us to study this case in depth.
Keywords: retinitis pigmentosa, sine pigmentum, unilateral.
Retinite pigmentosa sine pigmento unilateral: relato de caso
Resumo
Nosso objetivo é apresentar um caso de retinite pigmentosa sine pigmentum unilateral. Trata-se de uma paciente do sexo feminino, 41 anos, que foi avaliada após relatar diminuição da acuidade visual do olho direito (DO) há 2 anos. Ao exame físico, a acuidade visual no DO era 7/10, melhor corrigida, e no olho esquerdo, 10/10 sem correção. A biomicroscopia mostrou catarata subcapsular posterior estrelada no OD e no fundo dos olhos, estreitamento arteriolar, alteração do epitélio pigmentar da retina e lesões esbranquiçadas na região macular, também encontradas na periferia da retina. O campo visual computadorizado do OD mostrou padrão de contração, enquanto o eletrorretinograma indicou resposta subnormal sob condições escotópicas no OD. Por outro lado, os estudos realizados no olho esquerdo estavam dentro dos parâmetros normais. Concluindo, a apresentação de um caso de retinite pigmentosa sine pigmento unilateral é extremamente incomum, o que nos motivou a estudar este caso em profundidade.
Palavras-chave: retinite pigmentosa, sine pigmentum, unilateral.
Introducción
La retinosis pigmentaria (RP) es la distrofia retinal más frecuente con una prevalencia de 1:40001. Constituye un espectro heterogéneo de enfermedades degenerativas de la retina que pueden presentarse de modo esporádico o heredarse con un patrón autosómico dominante (AD), autosómico recesivo (AR) o ligado al cromosoma X (LXR).
La mayoría de los casos son monogénicos y la enfermedad es genéticamente heterogénea, con más de 70 genes mutados identificados. Los genes más comunes son rodopsina (RHO), responsable de aproximadamente el 25% de la RP dominante, usherin2A (USH2A), que causa alrededor del 20% de la enfermedad recesiva, y el regulador de la GTPasa de la retinosis pigmentaria (RPGR), responsable del 70% de la RP ligada al cromosoma X1.
Las características fundoscópicas características de la RP son la palidez del nervio óptico, vasos sanguíneos retinales atenuados y migración de pigmento intrarretinal (espículas óseas) en regiones de degeneración de fotorreceptores. Este pigmento en forma de espícula ósea corresponde a células que contienen melanina agrupadas alrededor de vasos sanguíneos ramificados en la retina interna. Estas células tendrían su origen luego de la muerte de fotorreceptores que provocaría la migración de las células del EPR hacia la retina interna con la formación de capas epiteliales notablemente polarizadas alrededor de los vasos sanguíneos retinales y contra la membrana limitante interna1.
La RP sine pigmento se clasifica como un subtipo atípico de retinitis que se distingue por la ausencia de cambios pigmentarios. Durante sus etapas iniciales, la deposición de pigmento puede variar desde escasa hasta abundante, lo que está condicionado por diversos factores como la edad del paciente, la duración de la enfermedad y una predisposición individual. Esta variabilidad puede interpretarse como un caso atípico.
Aunque la mayoría de los casos de RP son bilaterales, se han documentado casos en los que la afectación es unilateral. En la actualidad existen criterios para identificar un posible caso de RP unilateral, que son los siguientes: 1) presencia de cambios funcionales y una apariencia oftalmoscópica típica de RP en el ojo afectado; 2) ausencia de síntomas de RP en el otro ojo con un electrorretinograma (ERG) normal; 3) un período de observación prolongado (más de 5 años) para descartar un inicio tardío en el ojo no afectado; y 4) exclusión de causas inflamatorias en el ojo afectado2. El objetivo de este trabajo es exponer un caso atípico de RP unilateral sine pigmento, dos características sumamente atípicas en la presentación de la enfermedad.
Caso clínico
Paciente de 41 años evaluada en nuestra institución refiriendo disminución de la agudeza visual de ojo derecho (OD) de 2 años de evolución. No refirió antecedentes personales patológicos de relevancia.
Al examen físico, agudeza visual en OD 7/10 mejor corregida y en OI 10/10 SC. A la biomicroscopía, en OD se evidenciaba una catarata subcapsular posterior estrellada y en el fondo de ojos se constató estrechamiento arteriolar, alteración del EPR y lesiones blanquecinas en región macular que también se encontraban en la periferia retinal (fig. 1). El OI no mostraba alteraciones patológicas (fig. 2).
Posterior a estos hallazgos solicitamos estudios complementarios. El CVC del OD mostró un patrón de contracción (fig. 3), mientras que el ERG indicó una respuesta subnormal en condiciones escotópicas en el OD (fig. 4). Adicionalmente, se solicitó una retinofluoresceinografía (RFG) que evidenció hiperfluorescencia sin aumento en tiempos tardíos, conocido como efecto ventana (fig. 5).
La paciente fue controlada a los 2 años, donde se registró una AV de 2/10 mejor corregida en OD y 10/10 SC. No se observaron cambios en el fondo de ojo del OD en comparación con la última revisión.
En la actualidad, después de 10 años desde el diagnóstico inicial, la agudeza visual en el OD es de cuenta dedos a 30 cm, mientras que en el OI se mantiene en 10/10 sin corrección. La biomicroscopía del OD revela la presencia de una catarata nuclear y subcapsular posterior estrellada, mientras que el examen del fondo de ojos del OD muestra la persistencia de las lesiones descritas al momento del diagnóstico. No se observaron cambios patológicos en el OI.
Figura 1. Retinografía color del ojo derecho con estrechamiento arteriolar, alteración del epitelio pigmentario de la retina y lesiones blanquecinas en sector macular y periferia retinal.
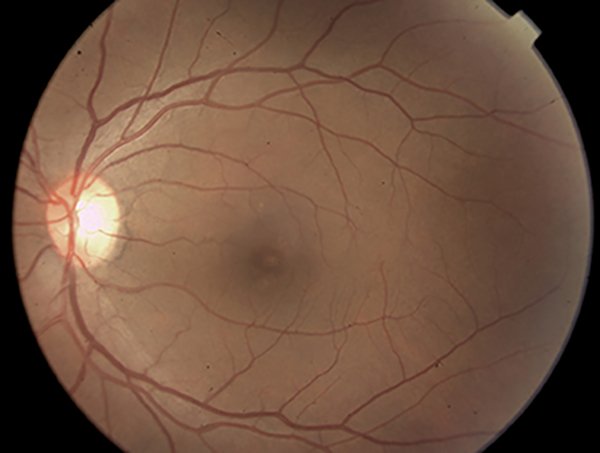
Figura 2. Retinografía color del ojo izquierdo sin cambios patológicos aparentes.
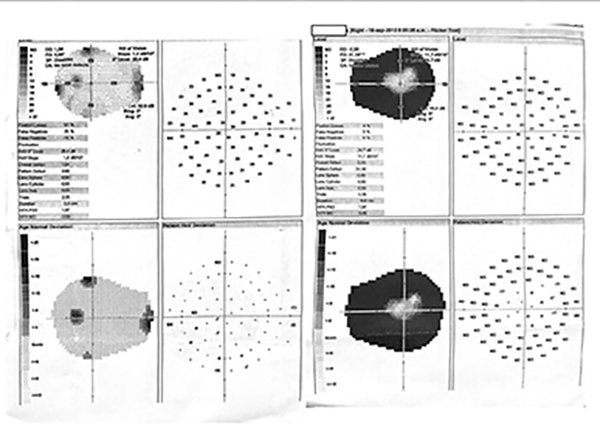
Figura 3. Campo visual computarizado de ojo derecho evidenciando patrón de contracción.
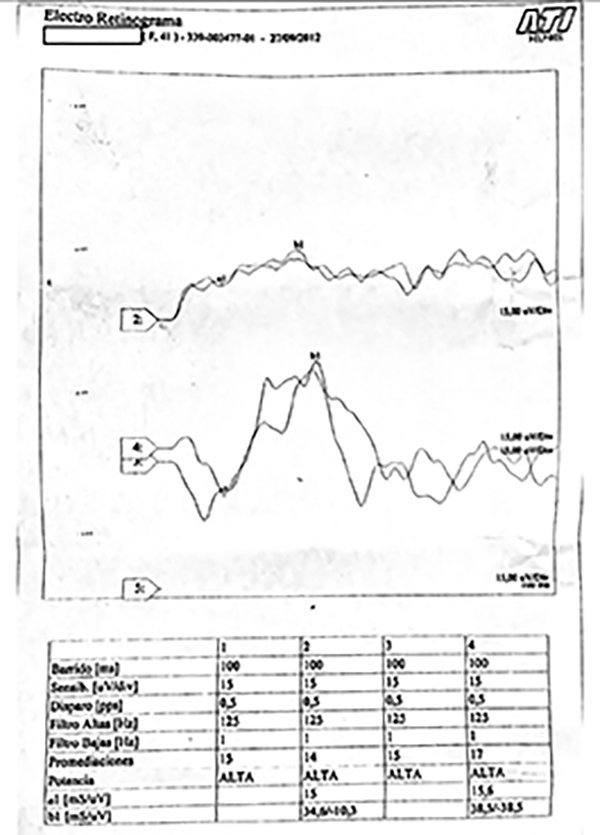
Figura 4. Electrorretinograma de ojo derecho con respuesta subnormal.
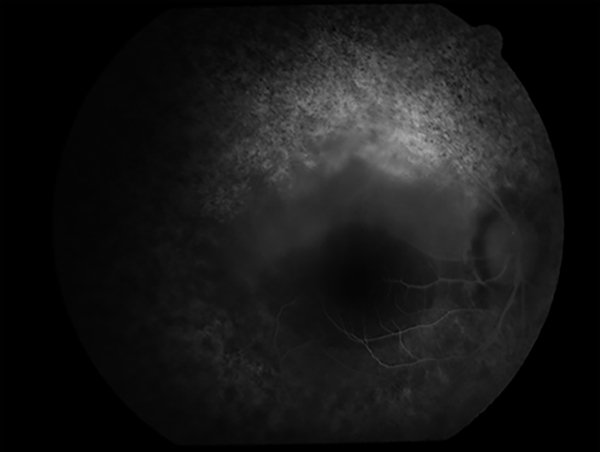
Figura 5. Retinofluoresceinografía de ojo derecho con hiperfluorescencia sin aumento en tiempos tardíos.
Discusión
La RP unilateral es una afección poco común, con una prevalencia estimada de 0,2% al 5% de todos los casos de RP, según diversas fuentes3-4. Para confirmar el diagnóstico se deben cumplir los criterios de François y Verriest, que incluyen la exclusión de todas las etiologías infecciosas, la presencia de signos clínicos de retinitis en el ojo afectado y la ausencia total de cualquier signo o síntoma de RP en el ojo contralateral2. La etiología de la RP unilateral aún no se ha esclarecido completamente y no hay pruebas concluyentes de que sea hereditaria5.
Por otro lado, la RP sine pigmento es una presentación atípica de esta enfermedad. Se cree que los cambios pigmentarios están influenciados por la edad de inicio, la duración de la enfermedad y la predisposición individual6. En el caso de nuestra paciente, presentaba ambas características, lo que la hace distintiva. El seguimiento a lo largo del tiempo —junto con la clínica y los estudios complementarios realizados— respaldan este diagnóstico, ya que en los controles posteriores mantuvo las mismas características sin afectación del OI.
Aunque un examen físico con evaluación fundoscópica puede ser muy sugerente en casos típicos de RP, en ocasiones donde la afectación es unilateral o el pigmento en la periferia retinal está ausente, son los métodos complementarios los que nos orientan hacia un diagnóstico preciso. En este caso, el ERG suele mostrar una reducción en la amplitud y un retraso en la respuesta de los bastones y conos7, mientras que la campimetría suele revelar un patrón de contracción.
Conclusión
La presentación inusual de la distrofia retinal más común genera un interés especial en estos casos. Es fundamental realizar una evaluación clínica exhaustiva y recurrir a estudios complementarios para lograr un diagnóstico preciso, dado que la pigmentación característica de la periferia de la retina puede desarrollarse gradualmente con el tiempo. En este contexto, se expuso el caso de un paciente con retinopatía pigmentaria unilateral donde se alienta a otros profesionales a considerar esta etiología en casos clínicos similares.
Referencias
1. Takahashi VKL, Takiuti JT, Jauregui R et al. Rates of bone spicule pigment appearance in patients with retinitis pigmentosa sine pigmento. Am J Ophthalmol 2018; 195: 176-180.
2. Francois J, Verriest G. Rétinopathie pigmentaire unilatérale. Ophthalmologica 1952; 124: 65-87.
3. Nazar C, Feldman M, González R, Espinoza R. Unilateral retinitis pigmentosa: a case report. Arch Soc Esp Oftalmol 2017; 92: 287-290.
4. Bhattarai D, Paudel N, Adhikari P et al. Unilateral retinitis pigmentosa. Nepal J Ophthalmol 2015; 7: 56-69.
5. Farrell DF. Unilateral retinitis pigmentosa and cone-rod dystrophy. Clin Ophthalmol 2009; 3: 263-270.
6. Pearlman JT, Saxton J, Hoffman G. Unilateral retinitis pigmentosa sine pigmento. Br J Ophthalmol 1976; 60: 354-360.
7. Sabbaghi H, Behbahani S, Daftarian N, Ahmadieh H. New criteria for evaluation of electroretinogram in patients with retinitis pigmentosa. Doc Ophthalmol 2021; 143: 271-281.