CASOS CLÍNICOS
Retinosis pigmentaria ligada al cromosoma X: reporte de caso clínico
Priscila Amado, Sebastián Amado
Instituto de Oftalmología Dr. Norberto Amado, Santa Fe, Argentina.
Oftalmol Clin Exp (ISSNe 1851-2658)
2024; 17(3): e408-e417.
DOI:10.70313/2718.7446.v17.n03.349
Resumen
Reportar un caso clínico de retinosis pigmentaria ligada al cromosoma X resaltando la importancia de realizar estudios complementarios y pruebas genéticas al paciente y su familia para identificar la mutación subyacente.
Se trata de un niño de 10 años que asiste a la consulta por cambios refractivos y dificultad para ver de noche (nictalopía). Como antecedente familiar relevante, la abuela materna presenta retinosis pigmentaria y la madre, miopía elevada (-14.0 D). Al examen oftalmológico presentaba una visión con corrección de 4/10 en ambos ojos, con una miopía de moderada a severa. En el fondo de ojo se observa una alteración pigmentaria moteada, difusa, con múltiples puntos blanco-amarillentos distribuidos en toda la media periferia, con atenuación arteriolar y leve palidez del nervio óptico bilateral. En la autofluorescencia se observó un anillo hiperautofluorescente alrededor de la mácula e hipoautofluorescencia más periférica. En la tomografía de coherencia óptica está preservada la zona elipsoide a nivel central y se afinan esas capas externas hacia la periferia. El electrorretinograma por flash mostró una disfunción tanto de conos como de bastones que no es absoluta. En el estudio genético se identificó una variante en hemicigosis en el gen RPGR, donde este resultado es compatible con retinosis pigmentaria ligada al cromosoma X.
En conclusión, es de suma relevancia que los pacientes en quienes se sospecha una distrofia de conos y bastones se estudien cuidadosamente mediante un examen oftalmológico completo, estudios complementarios, electrofisiológicos y fundamentalmente genéticos para identificar la mutación subyacente, ya que el advenimiento de la terapia génica podría brindarles una mejor calidad de vida.
Palabras clave: retinosis pigmentaria ligada al X, terapia génica, gen RPGR.
Cromosome X-linked pigmentary retinosis: case report
Abstract
To report a clinical case of X-linked Retinitis pigmentosa, highlighting the importance of complementary studies and genetic testing, to the patient and his family, to identify the underlying mutation. A 10-year-old boy was brought to the clinic for refractive changes and difficulty seeing at night (nyctalopia). As a relevant family history, the maternal grandmother had Retinitis pigmentosa and the mother had high myopia (14.0 D). On ophthalmologic examination she had 4/10 corrected vision in both eyes, with moderate to severe myopia. Fundus examination showed diffuse mottled pigmentary changes with multiple yellowish-white dots distributed throughout the midperiphery, with arteriolar attenuation and mild bilateral optic nerve pallor. Autofluorescence showed a hyperautofluorescent ring around the macula and more peripheral hypoautofluorescence. On optical coherence tomography the ellipsoid zone is preserved centrally and these outer layers are thinned towards the periphery. The flash electroretinogram showed a dysfunction of both cones and rods that is not absolute. Regarding the genetic study, a variant in hemizygosis has been identified in the RPGR gene, being this result compatible with X-linked retinitis pigmentosa.
In conclusion, it is of high relevance that those patients in whom we suspect a cone and rod dystrophy are extensively studied by a complete ophthalmologic examination, complementary studies, electrophysiologic and fundamentally genetic studies to identify the underlying mutation, since the advent of gene therapy could provide them with a better quality of life.
Keywords: X-linked retinitis pigmentosa, gene therapy, RPGR gene.
Retinite pigmentosa ligada ao cromossomo X: relato de caso clínico
Resumo
Relatar um caso clínico de retinite pigmentosa ligada ao cromossomo X, destacando a importância da realização de estudos complementares e exames genéticos no paciente e sua família para identificação da mutação subjacente.
Trata-se de um menino de 10 anos que procura a clínica devido a alterações refrativas e dificuldade de enxergar durate a noite (nictalopia). Como histórico familiar relevante, a avó materna tem retinite pigmentosa e a mãe tem alta miopia (-14,0 D). Ao exame oftalmológico apresentava visão corrigida para 4/10 em ambos os olhos, com miopia moderada a grave. No fundo observa-se alteração pigmentar difusa e mosqueada, com múltiplos pontos branco-amarelados distribuídos pela média periferia, com atenuação arteriolar e leve palidez do nervo óptico bilateral. A autofluorescência mostrou um anel hiperautofluorescente ao redor da mácula e hipoautofluorescência mais periférica. Na tomografia de coerência óptica, a zona elipsóide é preservada no nível central e essas camadas externas são afinadas em direção à periferia. O eletrorretinograma por flash mostrou uma disfunção de cones e bastonetes que não é absoluta. No estudo genético foi identificada uma variante hemizigótica no gene RPGR, onde esse resultado é compatível com retinite pigmentosa ligada ao cromossomo X.
Concluindo, é de extrema importância que os pacientes com suspeita de distrofia de cones-bastonetes sejam cuidadosamente estudados através de exame oftalmológico completo, estudos complementares, eletrofisiológicos e fundamentalmente genéticos para identificar a mutação subjacente, já que que o advento da terapia genética poderia lhes dar uma melhor qualidade de vida.
Palavras-chave: retinite pigmentosa ligada ao cromosoma X, terapia gênica, gene RPGR.
Introducción
La retinosis pigmentaria (RP) comprende un amplio grupo de enfermedades hereditarias caracterizadas por la pérdida primaria y progresiva de fotorreceptores y secundariamente de otras células retinales1. Se manifiesta inicialmente con ceguera nocturna y pérdida progresiva del campo visual, pero a menudo mantiene visión central hasta la etapa final de la enfermedad, aunque también suele manifestar visión borrosa, deslumbramiento y nistagmus2. Puede presentarse como una enfermedad que afecta exclusivamente al ojo (RP aislada o no sindrómica) o asociada a manifestaciones sistémicas (RP sindrómica: síndrome de Usher, de Bardet Biedl, enfermedad de Refsum, etc). Afecta a una de cada 3.000-5.000 personas en todo el mundo1. Se la considera como una enfermedad poco frecuente1-3.
Hay más de 100 variantes genéticas diferentes y se transmite siguiendo distintos patrones de herencia. Pueden heredarse de forma autosómica dominante, recesiva o ligada al X4. Los genes más comunes asociados con la retinitis pigmentaria son PRPF31, PRPH2, RDH12, RHO, RPE65, ABCA4, MAK, MERTK, NR2E3, PDE6B y RPGR1,4.
La RP ligada al cromosoma X es una de las formas más graves de degeneración de la retina y representa entre el 10% y el 20% de todos los casos de RP4. Hasta la fecha solo se han identificado 3 genes asociados con este patrón de herencia, donde el más común es el gen regulador de la GTPasa de la retinosis pigmentaria (RPGR)4. En estos casos, las mujeres transmiten la enfermedad, es decir son portadoras y los hombres la padecen. Sus hijos varones tienen un 50% de riesgo de padecer una RP y sus hijas tienen el mismo porcentaje de riesgo de ser portadoras heterocigotas sanas. Los hombres afectados nunca transmitirán una enfermedad ligada al cromosoma X a sus hijos; sin embargo, siempre lo harán sus cromosomas afectados a sus hijas, las que serán portadoras5-6.
Ante la sospecha clínica de RP (nictalopía, visión borrosa, deslumbramiento, nistagmus o constricción del campo visual) es de suma relevancia solicitar estudios complementarios, electrofisiológicos y fundamentalmente genéticos1-2, ya que incluso en la Argentina existe la posibilidad de realizar tratamientos en los casos adecuados, como fue planteado por Mancini y Saravia7, tema profundizado ampliamente en la revisión realizada por Ciccioli y Antacle8. Por lo pronto el propósito de este trabajo es resaltar la importancia de que todo paciente portador de una distrofia retinal debería ser secuenciado para obtener un diagnóstico correcto de la patología, conocer el gen afectado y la mutación para que esté informado y saber si es candidato o no a las terapias génicas en advenimiento.
Caso clínico
Paciente masculino de 10 años asiste a la consulta acompañado de su madre para control refractivo y por dificultad para ver de noche (nictalopía). Como antecedente familiar relevante, su abuela materna tiene retinosis pigmentaria y su madre, miopía elevada en ambos ojos (-14).
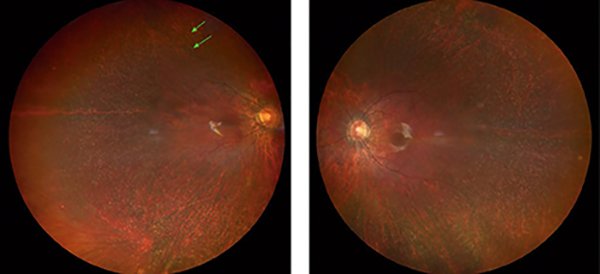
Al examen oftalmológico presenta: AV con corrección en OD 4/10 (-6 -0,75 x 15°) y OI 4/10 (-5 -100 x 140°). Examen biomicroscópico normal. En el fondo de ojo se observa una alteración pigmentaria moteada, difusa, con múltiples puntos blanco-amarillentos distribuidos en toda la media periferia, con atenuación arteriolar y leve palidez del nervio óptico bilateral (fig. 1).
La autofluorescencia (AF) revela un anillo hiperautofluorescente alrededor de la mácula e hipoautofluorescencia más periférica en ambos ojos (fig. 2). En el campo visual computado se observa un patrón de constricción periférica con remanente visual central (fig. 3) que coincide con la tomografía de coherencia óptica donde se preserva la zona elipsoide a nivel central, pero se van afinando esas capas externas hacia la periferia (fig. 4). En el ERG estandarizado por flash se utilizaron electrodos de contacto cutáneo que registró una disfunción moderada tanto de conos como de bastones. El electrooculograma (EOG) reveló afectación del epitelio pigmentario y el potencial visual evocado (PEV) fue normal. Debido a la alta sospecha de una distrofia de conos y bastones se derivó al paciente y a su familia a consejo genético. En él se ha identificado una variante en hemicigosis en el gen RPGR, donde este resultado fue compatible con retinosis pigmentaria ligada al cromosoma X. Por este motivo, se realizó el examen de fondo de ojo a la madre —que resultó normal—, pero la autofluorescencia mostraba un patrón de hiper e hipoautofluorescencia debido a la alteración del epitelio pigmentario en la media periferia, con conservación de éste a nivel central (fig. 5). En el fondo de ojo de la abuela se apreciaba un cuadro bien florido y la tríada típica de RP: presencia de osteoclastos o espículas óseas, atenuación arteriolar y palidez del nervio óptico (fig. 6), coincidiendo con lo observado en la AF (fig. 7).
Se realizó el diagnóstico diferencial con otras distrofias de conos y bastones, como la ceguera nocturna estacionaria congénita, la coroideremia, la atrofia girata, etc. Actualmente el paciente utiliza lentes aéreas de desenfoque (defocus) para enlentecer la progresión de su miopía, filtros para interiores de 450 nm y exteriores de 510 nm. Además, se realizó interconsulta con nutricionista para llevar una dieta antiinflamatoria con suplementos nutracéuticos y de esta manera tratar de preservar las células retinales lo más viables y sanas posible, mientras se espera el acceso a la terapia génica a la población general.
Figura 1. Fondo de ojo: alteración pigmentaria moteada, difusa, con múltiples puntos blanco-amarillentos distribuidos en la media periferia con atenuación arteriolar y leve palidez del nervio óptico bilateral.
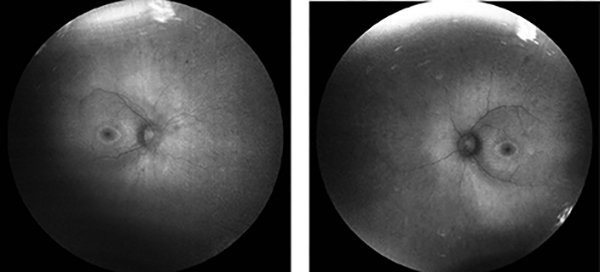
Figura 2. Autofluorescencia: obsérvese el anillo hiperautofluorescente alrededor de la mácula e hipoautofluorescencia más periférica en ambos ojos.
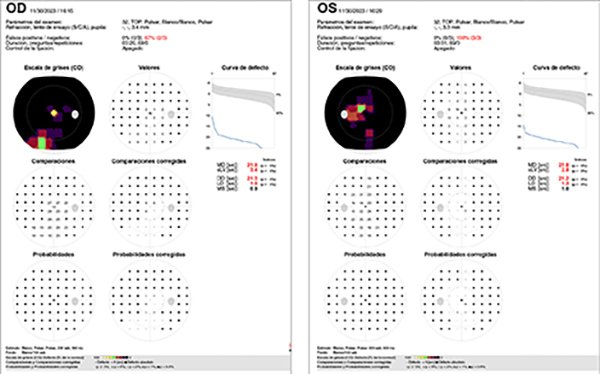
Figura 3. Campo visual computado: constricción periférica con remanente visual central.
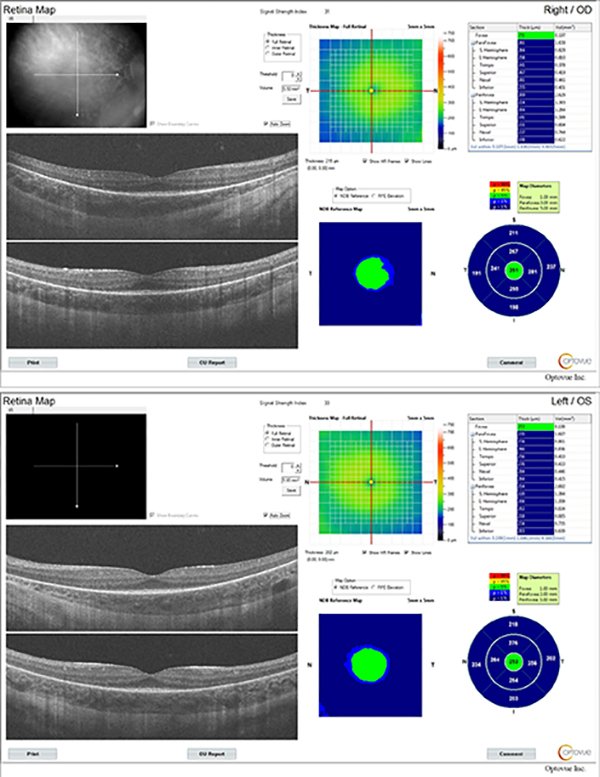
Figura 4. Tomografía de coherencia óptica: se preserva la zona elipsoide a nivel central, pero se van afinando las capas externas hacia la periferia.
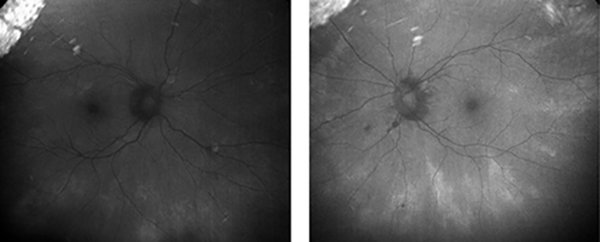
Figura 5. Autofluorescencia de la madre. Muestra un patrón de hiper e hipoautofluorescencia debido a la alteración del epitelio pigmentario en la media periferia con conservación de este a nivel central.
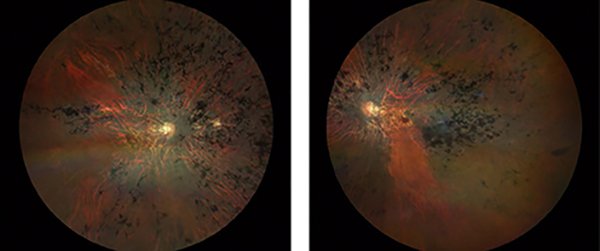
Figura 6. Fondo de ojo de la abuela. Se aprecia un cuadro bien florido y la tríada típica de RP: presencia de osteoclastos o espículas óseas, atenuación arteriolar y palidez del nervio óptico.
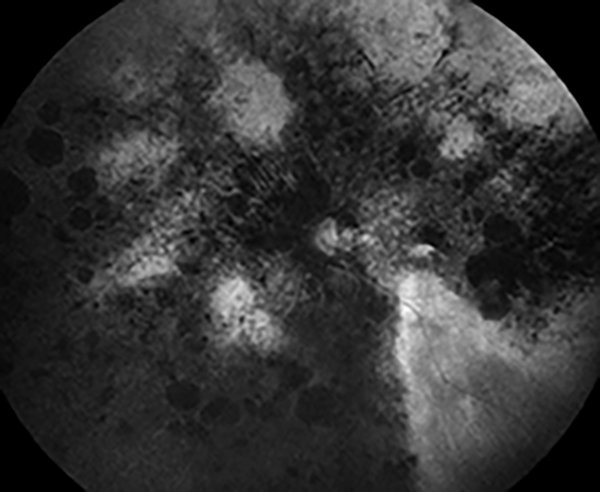
Figura 7. Autofluorescencia: presencia de hiper e hipoautofluorescencia difusa por compromiso del epitelio pigmentario.
Discusión
La retinosis pigmentaria ligada al cromosoma X (XLRP) es una forma grave de RP4,6, inicia tempranamente con nictalopía y puede progresar a ceguera legal en la tercera o cuarta década de la vida1-3. Mutaciones en el gen RPGR representan más del 70% de los casos de RP ligada al X4. La mayoría de los varones afectados tienen ceguera nocturna sintomática antes de los 10 años, suelen ser miopes y exhiben anomalías del fondo de ojo y cambios en el ERG en la primera infancia3. El examen de parientes femeninos cercanos es útil, ya que el reconocimiento del estado de portador confirmará el diagnóstico9.
Coincidiendo con la bibliografía revisada, nuestro paciente a sus 10 años ya presenta disminución de la visión, miopía severa y dificultad para la movilidad nocturna, por lo que se le han brindado herramientas para mejorar su independencia y su seguridad. Su madre —asintomática hasta el momento salvo por su miopía severa y desconociendo la patología— reveló en la AF el patrón típico de RP, al igual que su abuela.
El trípode más importante para arribar al diagnóstico clínico ha sido la AF, OCT y el ERG convencional por flash, los que también fueron de utilidad para hacer diagnósticos diferenciales y evaluar la progresión de la enfermedad10. Existen pruebas genéticas para la RP que son esenciales para obtener un diagnóstico preciso10. Conocer el gen mutado puede ayudar a una persona no solo a entender cómo la enfermedad puede afectar la visión durante su vida, sino también puede orientar a la realización de pruebas a los miembros de la familia para identificar a aquellos que corren riesgo de heredar la enfermedad. Además, conocer el defecto genético puede ayudar a nuestros pacientes a calificar para ensayos clínicos e informarles qué terapias futuras pueden ser beneficiosas7-8,11-14.
Actualmente hay varios ensayos clínicos prometedores en desarrollo para la terapia génica dirigida a la retinitis pigmentosa ligada al cromosoma X (XLRP) causada por mutaciones en el gen RPGR y algunas de ellas en fase 3, es decir que están en una etapa bastante avanzada, pero no podemos saber con certeza en qué momento estarán disponibles para la población general15-17. Estas terapias han informado mejoras en la sensibilidad retinal y/o agudeza visual. Consisten en la introducción de un gen correcto o anular alguno que no está funcionando bien o introducir un gen para que se produzca una función que la célula no tenía.
Como explicaron Ciccioli y Antacle en su revisión, estos tratamientos utilizan vectores virales adeno-asociados que no producen patogenicidad en seres humanos, exhiben un mínimo nivel de inmunogenicidad y transducen genes en un rango diverso de células blanco8. La degeneración celular en células retinales es irreversible, por lo tanto, la eficacia del vector está determinada por la habilidad de alcanzar efectivamente las células blanco y el mayor número de células viables posible para la inserción del gen. Por este motivo, es fundamental proteger a las células retinales mediante terapias no génicas como por ejemplo la nutricional18-20. Se trabaja sobre alimentos cuya función tiene que ver con la actividad antioxidante en órganos con una elevada producción de sustancias derivadas de procesos reactivos del oxígeno que pueden dañar las membranas celulares y facilitar la muerte celular, como es la retina. Otras herramientas pueden ser los filtros que mejoran la sensibilidad al contraste, eliminan los deslumbramientos y aumentan la luminosidad nocturna, así como también las ayudas ópticas de baja visión21-23.
Si bien actualmente aún no hay un tratamiento aprobado para la RP ligada al cromosoma X, el futuro es muy esperanzador ya que las características del gen RPGR cuyo tamaño le permite ser empaquetado en vectores virales adeno-asociados han despertado un gran interés entre la comunidad científica y clínica en los últimos años y probablemente lo tengamos disponible para la población general en un futuro no muy lejano.
Conclusión
En la actualidad se encuentran en desarrollo protocolos de nuevas terapias para enfermedades como la amaurosis congénita de Leber, la enfermedad de Stargardt, el síndrome de Usher, la retinosis pigmentaria por mutaciones en el gen RPGR, la coroideremia y muchas más.
Por lo tanto, es de suma relevancia que aquellos pacientes en quienes sospechamos una distrofia de conos y bastones sean ampliamente estudiados mediante un examen oftalmológico completo, estudios complementarios, electrofisiológicos y fundamentalmente genéticos para identificar la mutación subyacente, ya que el advenimiento de la terapia génica ofrece el potencial de detener la pérdida progresiva de la visión y del campo visual, lo que puede mejorar la visión, la sensibilidad a la luz, el contraste y de esta manera brindarle al paciente una mejor calidad de vida.
Referencias
1. Vingolo EM, Mascolo S, Miccichè F, Manco G. Retinitis pigmentosa: from pathomolecular mechanisms to therapeutic strategies. Medicina (Kaunas) 2024; 60: 189.
2. Tsang SH, Sharma T. Retinitis pigmentosa (non-syndromic). Adv Exp Med Biol 2018; 1085: 125-130.
3. Chang S, Vaccarella L, Olatunji S et al. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Curr Genomics 2011; 12: 267-275.
4. Liu W, Liu S, Li P, Yao K. Retinitis pigmentosa: progress in molecular pathology and biotherapeutical strategies. Int J Mol Sci 2022; 23: 4883.
5. Motta FL, Martin RP, Filippelli-Silva R et al. Relative frequency of inherited retinal dystrophies in Brazil. Sci Rep 2018; 8: 15939.
6. Lyraki R, Megaw R, Hurd T. Disease mechanisms of X-linked retinitis pigmentosa due to RP2 and RPGR mutations. Biochem Soc Trans 2016; 44: 1235-1244.
7. Mancini JE, Saravia M. Terapia génica en la Argentina, ¿es posible?: un elemento más para el tratamiento de enfermedades raras de la retina. Oftalmol Clin Exp 2023; 16: e342-e345.
8. Ciccioli M, Antacle A. Revisión sobre terapia génica aprobada y recuperación visual en la amaurosis congénita de Leber por mutación en el gen RPE65. Oftalmol Clin Exp 2024; 17: e33-e40.
9. Tran M, Kolesnikova M, Kim AH et al. Clinical characteristics of high myopia in female carriers of pathogenic RPGR mutations: a case series and review of the literature. Ophthalmic Genet 2023; 44: 295-303.
10. Fahim A. Retinitis pigmentosa: recent advances and future directions in diagnosis and management. Curr Opin Pediatr 2018; 30: 725-733.
11. Cehajic-Kapetanovic J, Xue K, Martínez-Fernández de la Cámara C et al. Initial results from a first-in-human gene therapy trial on X-linked retinitis pigmentosa caused by mutations in RPGR. Nat Med 2020; 26: 354-359.
12. von Krusenstiern L, Liu J, XIRIUS Part 1 Study GroupXOLARIS Study Group et al. changes in retinal sensitivity associated with cotoretigene toliparvovec in X-linked retinitis pigmentosa with RPGR gene variations. JAMA Ophthalmol 2023; 141: 275-283.
13. Lam BL, Pennesi ME, XIRIUS Study Group et al. Assessment of visual function with cotoretigene toliparvovec in x-linked retinitis pigmentosa in the randomized XIRIUS phase 2/3 Study. Ophthalmology 2024; 131: 1083-1093.
14. Ikeda HO, Hasegawa T, Abe H et al. Efficacy and safety of branched chain amino acids on retinitis pigmentosa: a randomized, double-blind, placebo-controlled clinical trial. Transl Vis Sci Technol 2024; 13: 29.
15. Stansfield N. Gene therapy for X-linked retinitis pigmentosa shows safety, efficacy in phase 1/2 study [en línea]. Cranbury, NJ: CGTlive, 2022. Disponible en: https://www.cgtlive.com/view/gene-therapy-for-x-linked-retinitis-pigmentosa-shows-safety-efficacy-in-phase-1-2-study
16. Beacon Therapeutics reports encouraging interim results from phase 2 clinical trial for XLRP gene therapy [en línea]. Columbia MD: Foundation Fighting Blindness, Feb 2, 2024. Disponible en: https://www.fightingblindness.org/news/beacon-therapeutics-reports-encouraging-interim-results-from-phase-2-clinical-trial-for-xlrp-gene-therapy-869
17. Beacon doses first patient in its phase 2/3 VISTA clinical trial for XLRP gene therapy [en línea]. Columbia MD: Foundation Fighting Blindness, June 13, 2024. Disponible en: https://www.fightingblindness.org/news/beacon-doses-first-patient-in-its-phase-2-3-vista-clinical-trial-for-xlrp-gene-therapy-897
18. Olivares-González L, Salom D, González-García E et al. NUTRARET: effect of 2-year nutraceutical supplementation on Redox status and visual function of patients with retinitis pigmentosa: a randomized, double-blind, placebo-controlled trial. Front Nutr 2022; 9: 847910.
19. Comander J, Weigel DiFranco C, Sanderson K et al. Natural history of retinitis pigmentosa based on genotype, vitamin A/E supplementation, and an electroretinogram biomarker. JCI Insight 2023; 8: e167546.
20. Neelam K, Dey S, Sim R et al. Fructus lycii: a natural dietary supplement for amelioration of retinal diseases. Nutrients 2021; 13: 246.
21. Cedrún-Sánchez JE, Chamorro E, Bonnin-Arias C et al. Visual discrimination increase by yellow filters in retinitis pigmentosa. Optom Vis Sci 2016; 93: 1537-1544.
22. Pastor-Idoate S, Mateos-Olivares M, Sobas EM et al. Short-wavelength light-blocking filters and oral melatonin administration in patients with retinitis pigmentosa: protocol for a randomized controlled trial. JMIR Res Protoc 2023; 12: e49196.
23. Nguyen XT, Koopman J, van Genderen MM et al. Artificial vision: the effectiveness of the OrCam in patients with advanced inherited retinal dystrophies. Acta Ophthalmol 2022; 100: e986-e993.