CASOS CLÍNICOS
Síndrome de Sotos, afección corneal y escleral: reporte de caso
Rubén G. Záratea, Verónica A. Bebigliaa, Rocío Mendezb, Carla Custoc, Carlos Contrerasd, Gustavo H. Di Rienzoe, Alina Laura Zabalaf, Gretel Rauschg
aServicio de Oftalmología, Hospital Regional Río Grande, Tierra del Fuego, Argentina.
bServicio de Clínica Médica, Hospital Regional Río Grande, Tierra del Fuego, Argentina.
cServicio de Endocrinología, Hospital Regional Río Grande, Tierra del Fuego, Argentina.
dServicio de Diagnóstico por Imágenes, Hospital Regional Río Grande, Tierra del Fuego, Argentina.
eCentro de Imágenes Médicas (CIM), Río Grande, Tierra del Fuego, Argentina.
fServicio de Neurología, Hospital Regional Río Grande, Tierra del Fuego, Argentina.
g Consultorio Privado de Reumatología, Río Grande, Tierra del Fuego, Argentina.
Oftalmol Clin Exp (ISSNe 1851-2658)
2024; 17(1): eXX-eXX.
Agradecimientos
A María José Ledesma, Cintia Calixto, Johana V. Díaz, Lucas Daniel Moreno, por su colaboración para el manejo del presente caso clínico.
Resumen
El objetivo es describir las alteraciones oftalmológicas, en especial de superficie ocular, que permitieron generar la sospecha clínica y posterior diagnóstico y manejo terapéutico interdisciplinario de una paciente con síndrome de Sotos.
Una mujer de 25 años consultó por fotofobia, ardor y dolor de varios meses de evolución. A la biomicroscopía se observaron infiltrados corneales periféricos e inflamación límbica bilateral. Se realizó anamnesis para evaluar alteraciones generales relacionadas con ojo seco en mujer joven. Se destacó la ausencia de menstruación, alteraciones cardiológicas, mandibulares y agenesia renal, además de signos físicos llamativos. Por estos datos se comenzó a sospechar una patología sistémica. Se realizaron interconsultas inicialmente con especialistas en reumatología, endocrinología y se confirmó posteriormente el diagnóstico de síndrome de Sotos. Se realizó tratamiento tópico con lubricantes e inmunomodulares junto con la evaluación médica de otras especialidades, logrando el control terapéutico del cuadro.
El síndrome de Sotos es una patología genética de muy baja prevalencia que puede presentarse con signos oculares. En este caso se ha descrito la potencial asociación a queratitis periférica con reacción limbar. Se destaca la relevancia que de la anamnesis y el trabajo interdisciplinario, tanto para establecer el diagnóstico como su posterior manejo terapéutico.
Palabras clave: síndrome de Sotos, queratitis, queratitis periférica, ojo seco, superficie ocular, escleritis, escleromalacia, infiltrados limbares.
Sotos syndrome, corneal and scleral involvement: case report
Abstract
The objective is to describe the ophthalmologic alterations, especially of the ocular surface, that led to the clinical suspicion and subsequent diagnosis and interdisciplinary therapeutic management of a patient with Sotos syndrome.
A 25-year-old woman consulted for photophobia, burning and pain of several months of evolution. Biomicroscopy showed peripheral corneal infiltrates and bilateral limbal inflammation. Anamnesis was performed to evaluate general alterations related to dry eye in a young woman. The absence of menstruation, cardiac and mandibular alterations and renal agenesis were highlighted, in addition to remarkable physical signs. Initial consultations were made with specialists in rheumatology and endocrinology, later confirming the diagnosis of Sotos syndrome. Topical treatment with lubricants and immunomodulators was performed, together with medical evaluation of other specialties, achieving therapeutic control of the condition.
Sotos syndrome is a genetic pathology of very low prevalence, which can present with ophthalmologic signs. In this case, the potential association to peripheral keratitis with limbar reaction has been described. The relevance of anamnesis and interdisciplinary work to establish the diagnosis and subsequent therapeutic management is emphasized.
Keywords: Sotos syndrome, keratitis, peripheral keratitis, dry eye, ocular Surface, scleritis, scleromalacia, limbal infiltrates.
Síndrome de Sotos, afecção corneana e escleral: relato de caso
Resumo
O objetivo é descrever as alterações oftalmológicas, principalmente da superfície ocular, que permitiram a geração de suspeita clínica e posterior diagnóstico e manejo terapêutico interdisciplinar de um paciente com síndrome de Sotos.
Uma mulher de 25 anos apresentou fotofobia, queimação e dor que persistiam há vários meses. A biomicroscopia revelou infiltrados corneanos periféricos e inflamação límbica bilateral. A anamnese foi coletada para avaliar alterações gerais relacionadas ao olho seco em uma mulher jovem. Destacaram-se a ausência de menstruação, alterações cardiológicas e mandibulares e agenesia renal, além de sinais físicos marcantes. Com base nesses dados, começou-se a suspeitar de uma patologia sistêmica. Inicialmente foram feitas consultas com especialistas em reumatologia e endocrinologia e posteriormente confirmado o diagnóstico de síndrome de Sotos. O tratamento tópico com lubrificantes e imunomoduladores foi realizado juntamente com avaliação médica de outras especialidades, obtendo controle terapêutico do quadro.
A síndrome de Sotos é uma patologia genética de prevalência muito baixa que pode apresentar sinais oculares. Neste caso, foi descrita a potencial associação de ceratite periférica com reação do limbo. Destaca-se a relevância da anamnese e do trabalho interdisciplinar, tanto para o estabelecimento do diagnóstico quanto para seu posterior manejo terapêutico.
Palavras-chave: Síndrome de Sotos, ceratite, ceratite periférica, olho seco, superfície ocular, esclerite, escleromalácia, infiltrados limbais.
Introducción
El síndrome de Sotos (SS) es una enfermedad genética con un patrón de herencia autosómico dominante causado por haploinsuficiencia del gen NSD1 secundaria a mutaciones puntuales o microdeleciones del locus 5q35 en el que está ubicado el gen1. Se ha postulado la hipótesis de que la alteración radica en la expresión de una proteína presente en el cerebro, el musculo esquelético, el riñón, el bazo, el timo y el pulmón1-2. Aunque tal función biológica aún no ha sido establecida con exactitud, se propone una relación con genes que promueven el crecimiento1-2.
Este síndrome, con incidencia en 7 por cada 100.000 nacimientos3-4, se caracteriza por presentar un crecimiento prenatal y posnatal excesivo, con macrocefalia, facies específicas con paladar alto, mandíbula prominente, línea de pelo fronto-temporal elevada, fisuras palpebrales, enrojecimiento malar, paladar ojival o arqueado, hipertelorismo aparente, cara larga, mentón prominente, pabellones auriculares grandes3. También se manifiesta con alteraciones odontológicas como erupción prematura de los dientes o amelogénesis imperfecta3. El retraso mental que varía de leve a grave está presente en el 80% de los casos con dificultad en el procesamiento verbal, el razonamiento abstracto y la escritura, donde además los pacientes pueden tener convulsiones, alteraciones conductuales y trastornos psiquiátricos, como aislamiento social, agresividad, depresión, ansiedad y psicosis5. Las anomalías cardíacas, presentes en el 25% de los casos, pueden ser comunicación interauricular, interventricular, ductus arterioso persistente y estenosis pulmonar y tricúspide. Con menor frecuencia se evidencian anomalías renales con reflujo vesicouretral e infecciones urinarias recurrentes, otitis media a repetición.
Estos pacientes también tienen infecciones respiratorias de repetición, escoliosis, reflujo gastroesofágico, hernias inguinales y alteraciones endocrinológicas como hipotiroidismo primario6. Otro hallazgo informado en la literatura es el riesgo aumentado de neoplasias (neuroblastoma, teratoma sacrococcígeo, leucemia linfoblástica aguda, tumor de Wilms6. Se han reportado también alteraciones oftalmológicas tales como estrabismo, cataratas y atrofia macular, además del hipertelorismo aparente, displasia septo-óptica, megaoftalmos, palidez del disco óptico, cataratas, estrabismo, nistagmus3. Pero hasta el presente no se han encontrado informes que postulen la potencial presentación de alteraciones de la superficie ocular en el contexto del síndrome de Sotos. Por lo tanto, este estudio propone describir un caso clínico de una mujer joven con alteraciones de córnea y esclera, cuyo diagnóstico de síndrome de Sotos y el posterior manejo clínico interdisciplinario se logró a partir del control oftalmológico.
Caso clínico
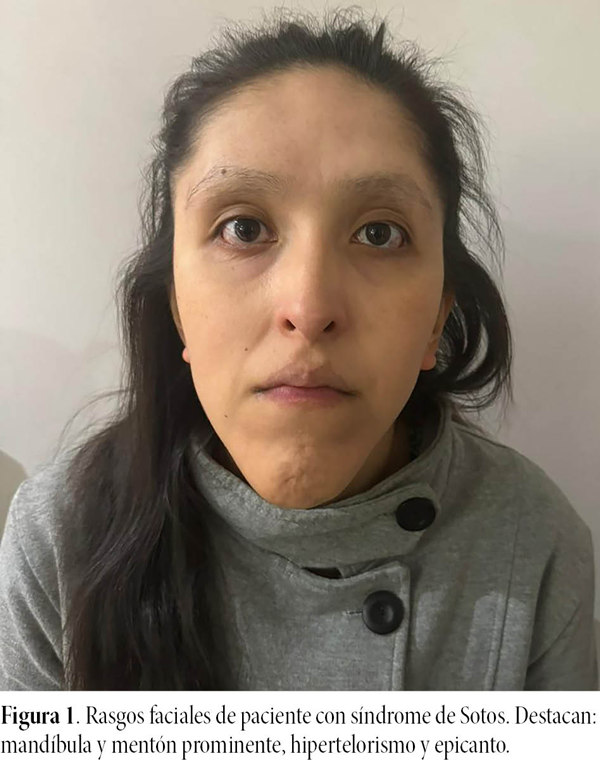
Paciente de 25 años de sexo femenino que concurre al Servicio de Oftalmología el 4 de mayo de 2023, acompañada de su madre, por dolor binocular, ardor e imposibilidad de apertura palpebral. Al realizar la anamnesis sobre antecedentes generales, en el interrogatorio la madre refiere que su hija presenta amenorrea continua de larga evolución sin presentar algún tratamiento sistémico sobre ella y también tiene antecedentes cardiológicos (cirugía interauricular). Otro dato relevante es que refirió agenesia del riñón izquierdo. También, la paciente tenía rasgos faciales particulares (fig. 1), como mandíbula y mentón prominente, hipertelorismo y epicanto.
En el momento de la consulta presentaba una agudeza visual disminuida 6 décimas en ambos ojos. A la biomicroscopía se observaron infiltrados corneales limbares bilaterales (fig. 2), progresión de vasos esclerales a córnea (fig. 3), adelgazamiento escleral leve con queratitis periférica superficial, secreción serosa de ambos ojos y un tiempo de ruptura lagrimal o break up time (BUT) disminuido (entre 3 a 4 segundos). Tenía presión intraocular en ambos ojos de 12 mmHg y en el fondo de ojo presentaba retina aplicada en sus cuatro cuadrantes, vítreo libre, mácula y nervio óptico sin particularidades con un recorrido vascular normal. Se interpretó de forma inicial problema de superficie ocular relacionado con enfermedad sistémica. Se comenzó tratamiento con ciclosporina tópica 0,005 cada 12 horas), loteprednol 0,5 mg (cada 8 horas), olopatadina 0,1 mg tópica (cada 12 horas) y un lubricante ocular a base de hialuronato de sodio 0,4 mg libre de conservantes. Se explicó a la paciente cuadro de ojo seco cuyo origen debía estudiarse y se solicitó interconsulta con el servicio de ginecología, el área de reumatología y clínica médica, endocrinología y cardiología, para evaluación interdisciplinaria debido a mujer joven con síndrome de ojo seco en contexto de una potencial enfermedad general, dadas las características físicas y los antecedentes clínicos referidos. Se citó para realizar un seguimiento a control en 7 días.


La paciente regresó a control a los 12 días de su primera evaluación con notable mejoría. Su agudeza visual aumentó a 8 décimas y a la biomicroscopía presentó una disminución en la congestión conjuntival junto con una disminución en la secreción serosa. Se realizó topografía corneal y tomografía de coherencia óptica (OCT) que no arrojaron ninguna alteración relevante. La madre refirió que la paciente comenzó con terapia hormonal indicada por el servicio endocrinológico por habérsele detectado una agenesia ovárica. Tras haber descartado otras causas de ojo seco sistémico y por los datos clínicos generales y las características físicas de la paciente, luego de interactuar entre oftalmología y los otros servicios, se realizó el diagnóstico de síndrome de Sotos, explicado a la paciente y a sus familiares.
Transcurrido un mes, la paciente presentó mejoría notable a la biomicroscopía, observándose una disminución del infiltrado corneal y retroceso casi por completo de los vasos limbares con ausencia de secreción. Además, su agudeza visual siguió mejorando, logrando en ojo derecho del 10 décimas y 9 décimas del ojo izquierdo, con evidencia también de una mejoría emocional importante. Se le recomendó continuar con igual plan terapéutico y control en un mes.
A los 35 días la paciente regresó con dolor binocular, leve secreción serosa, molestias referidas como pinchazos y dolor (la madre dijo que por motivos económicos no pudo acceder a la medicación completa). Se puso en contacto a la paciente con servicios sociales de la institución y se indicó retomar el tratamiento realizado en la primera consulta. Se vuelve a citar a control en un mes.
La paciente regresó con estudios complementarios donde se observaron en la resonancia magnética nuclear (RMN) de cerebro con contraste, múltiples focos hiperintensos en secuencias de T2 y FLAIR en sustancia blanca frontal y subcorticales de forma bilateral, de carácter inespecífico (fig. 4). En la última consulta realizada a fines de diciembre de 2023 (al mes del control anterior), la paciente se encontró con notable mejoría y nuevamente se le indicó mantener seguimiento y control mensual con disminución del loteprednol (cada 12 horas), habiéndose constatado que la presión intraocular se mantenía estable (12 mmHg en ambos ojos) y el fondo de ojos seguía normal.

Discusión
En este trabajo se describió un caso de síndrome de Sotos cuyo diagnóstico y posterior manejo terapéutico fue logrado a partir de la consulta oftalmológica. Para poder establecer el diagnóstico de esta patología1, 3 se postularon cuatro criterios diagnósticos como los más relevantes: apariencia facial característica, crecimiento excesivo y acelerado con talla, perímetro cefálico, manos y pies por encima del percentil 97, edad ósea avanzada y retraso en el desarrollo psicomotor. A su vez, el diagnóstico diferencial del síndrome de Sotos debe hacerse con otros síndromes de sobrecrecimiento, entre ellos el más importante es el síndrome X frágil7. La prevalencia de este síndrome es mayor que la reportada en el Sotos; se estima en 1 en 5.000 hombres y 1 en 4.000 a 6.000 mujeres. Es la segunda causa de retraso mental con origen genético después del síndrome de Down y su fenotipo es más evidente en hombres que en mujeres. Presenta talla elevada, macrocefalia, cara alargada, mentón prominente, frente alta, laxitud ligamentosa y macro-orquidismo de aparición en la pubertad1,7.
Cabe destacar que la suma de características que pueden o deberían hacer pensar en este síndrome están relacionadas con la observación macroscópica que debe hacer el médico, incluso dentro del contexto de la consulta oftalmológica para poder así sospechar la patología. Tenemos por ejemplo la cara alargada con línea del pelo elevada, orejas prominentes y además su asociación con retraso madurativo con leve retraso mental. En el caso presentado, la paciente tenía otros datos muy sugestivos del síndrome de Sotos: los antecedentes de cirugía cardíaca (de comunicación interauricular), atresia renal izquierda, huesos largos y aparente hipertelorismo, compromiso hormonal por atresia ovárica. Por las características del caso se evaluó la posibilidad de un diagnóstico diferencial con acromegalia, pero luego, al considerar el resto de manifestaciones, permitió la confirmación clínica de síndrome de Sotos.
Este caso tiene una manifestación florida de signos, destacando también el hallazgo de las lesiones cerebrales. Creemos que esto puede estar en contexto con una alteración neurovascular, algo que está actualmente en seguimiento y evaluación por el servicio de neurología.
A nivel oftalmológico la literatura describe hipertelorismo aparente, displasia septo-óptica, megaoftalmos, palidez del disco óptico, cataratas, estrabismo y nistagmos3. Como aporte original de este estudio, en particular se encontraron en la biomicroscopía infiltrados corneales, queratitis periférica leve y adelgazamiento escleral, que no habían sido comunicados previamente en la literatura asociados a este tipo de enfermedad. Si bien nuestro trabajo no alcanza a expresar una directa relación, se pretende aportar esta observación a la comunidad científica para que pueda considerarse que las alteraciones de la superficie ocular deben ser exploradas y evaluadas ante pacientes con sospecha diagnóstica de síndrome de Sotos.
Como limitación, cabe mencionar que hubiera sido ideal tener las imágenes de la superficie ocular del primer día de consulta, cuando los signos eran más severos, pero por la fotofobia de la paciente no se pudieron tomar ni tampoco existía la sospecha inicial de la relevancia de todo el cuadro clínico que se consideró posteriormente. Además, otros estudios de mayor resolución como podría ser una microscopía confocal hubiese sido ideal para caracterizar esta afectación, pero no teníamos la posibilidad de realizarlo en nuestra región y tampoco era factible solicitarle a la paciente que viaje para hacerlo en otro lugar, como la ciudad de Buenos Aires, por cuestiones de costos y lejanía geográfica.
En cuanto a la evolución, en nuestro caso la alteración de la superficie ocular respondió de manera favorable con el tratamiento tópico oftalmológico, pero no podemos descartar que eventualmente podría también presentar una mejoría asociada al tratamiento concomitante hormonal realizado por vía oral. Se pueden hacer hipótesis sobre diferentes mecanismos que podrían relacionar el síndrome de Sotos con la afectación de superficie ocular observada, que característicamente afectó el área periférica de la córnea y el limbo, como suele observarse en patologías de autoinmunes, pero también está el contexto endócrino que posiblemente también pueda tener alguna relación. Esperamos que el presente trabajo estimule la realización de futuros estudios que clarifiquen estos aspectos.
Conclusión
El presente trabajo describió la manifestación oftalmológica en general y la afectación de la superficie ocular en particular en el contexto de una paciente con síndrome de Sotos. Al ser una anomalía de baja prevalencia, se resalta la importancia de comunicar esta forma de presentación con queratitis periférica y reacción limbar. Se destaca que —como en este caso— es el médico oftalmólogo quien puede generar la sospecha diagnóstica del síndrome de Sotos y mediante el trabajo interdisciplinario lograr su posterior confirmación y el adecuado manejo terapéutico.
Referencias
1. Lapunzina P. Síndrome de Sotos. Protoc Diagn Ter Pediatr 2010; 1: 71-79. Disponible en: https://www.researchgate.net/profile/Pablo-Lapunzina/publication/265820588_SINDROME_DE_SOTOS/links/54ef89550cf2432ba656e5d8/SINDROME-DE-SOTOS.pdf
2. Fagali C, Kok F, Nicola P et al. MLPA analysis in 30 Sotos syndrome patients revealed one total NSD1 deletion and two partial deletions not previously reported. Eur J Med Genet 2009; 52: 333-336.
3. Baujat G, Cormier-Daire V. Sotos syndrome. Orphanet J Rare Dis 2007; 2: 36.
4. Root AW, Diamond FB. The Sotos syndrome-NSD1 haploinsufficiency: cerebral gigantism update. Growth Genet Horm 2006; 22: 33-38.
5. McKusick VA, Rasmussen SA. Sotos syndrome 1. En: Online Mendelian Inheritance in Man (OMIM). Baltimore, US: McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University School of Medicine, 2022: entrada #117550. Disponible en: http://omim.org/entry/117550
6. Tatton-Brown K, Cole TRP, Rahman N et al. Sotos syndrome. GeneReviews [en línea]. Última revisión: 1 dic. 2022. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1479/#sotos
7. Saldarriaga W, Tassone F, González-Teshima LY et al. Fragile X syndrome. Colomb Med (Cali) 2014; 45: 190-198.