CLINICAL CASES
Coexistence of myotonic muscular dystrophy and Fuchs corneal endothelial dystrophy: case report
Rocío A. Gomez, Tomás Etcheverry, Laura E. Alba, Alejandro Signorelli, Ricardo Zaldúa, Gustavo J. Galperín
Centro Médico Oftalmológico IOFA, Buenos Aires, Argentina.
Oftalmol Clin Exp (ISSNe 1851-2658)
2024; 17(1): eXX-eXX.
Abstract
We present the case of a 41-year-old woman with an established diagnosis of Steinert’s myotonic dystrophy and the presence of signs compatible with Fuchs endothelial corneal dystrophy. In patients with Steinert there may be an association with Fuch’s as a cause of corneal edema. Therefore, it is essential to carry out periodic ophthalmological controls. On the other hand, considering the association of type 1 Steinert’s myotonic dystrophy with the development of cataract, it is suggested specular microscopy be performed before carrying out any surgical procedure in order to evaluate the corneal endothelium prior to the intervention.
Keywords: corneal disease, Fuchs endothelial corneal dystrophy, motonic dystrophy, corneal edema.
Coexistencia de distrofia miotónica y distrofia corneal endotelial de Fuchs: informe de caso
Resumen
Reportar el caso de una paciente de 41 años de edad con diagnóstico previo de distrofia miotónica de Steinert y presencia de signos compatibles con distrofia corneal endotelial de Fuchs. En pacientes con distrofia miotónica puede haber una asociación con la de Fuchs como causa de edema corneal. Por ello, es fundamental realizar controles oftalmológicos periódicos. Por otro lado, considerando la asociación de la distrofia miotónica de Steinert tipo 1 con el desarrollo de cataratas, se sugiere realizar microscopía especular antes de hacer cualquier procedimiento quirúrgico con el fin de evaluar el endotelio corneal previo a la intervención.
Palabras clave: distrofia corneal endotelial de Fuchs, distrofia miotónica, patología corneal, edema corneal.
Coexistência de distrofia miotônica e distrofia endotelial corneana de Fuchs: relato de caso
Resumo
Relatar o caso de um paciente de 41 anos com diagnóstico prévio de distrofia miotônica de Steinert e presença de sinais compatíveis com distrofia endotelial corneana de Fuchs. Em pacientes com distrofia miotônica pode haver associação com a distrofia de Fuchs como causa de edema corneano. Portanto, é fundamental realizar exames oftalmológicos regulares. Por outro lado, considerando a associação da distrofia miotônica tipo 1 de Steinert com o desenvolvimento de catarata, sugere-se a realização de microscopia especular antes de realizar qualquer procedimento cirúrgico para avaliar o endotélio corneano antes da intervenção.
Palavras-chave: Distrofia corneana endotelial de Fuchs, distrofia miotônica, patologia corneana, edema corneano.
Introduction
Myotonic dystrophy (MD) is a slowly progressive chronic disease with autosomal dominant inheritance, characterized by marked intra and interfamilial clinical variability1. It is a syndrome with multisystem affection that includes myotonia, muscular dystrophy, cardiac conduction defects, endocrine and ophthalmological disorders2. Regarding the muscle compromise, the detection of typical myotonic discharges with small amplitude polyphasic potentials is characteristic in the electromyogram3. The medical literature describes frequent ophthalmological findings in association. Our purpose is to report a case of a patient with an established diagnosis of Steinert’s myotonic dystrophy and the presence of signs compatible with Fuchs endothelial corneal dystrophy (FECD).
Methods
We present the case of a 41-year-old woman who attended the emergency room (ER) for sudden decrease in visual acuity on the left eye. As a relevant pathological antecedent, the patient had a diagnosis of type 1 Steinert’s myotonic dystrophy (MD1), confirmed by a genetic study carried out 25 years earlier and has been on intermittent treatment with modafinil for six years at a dose of 200 mg per day.
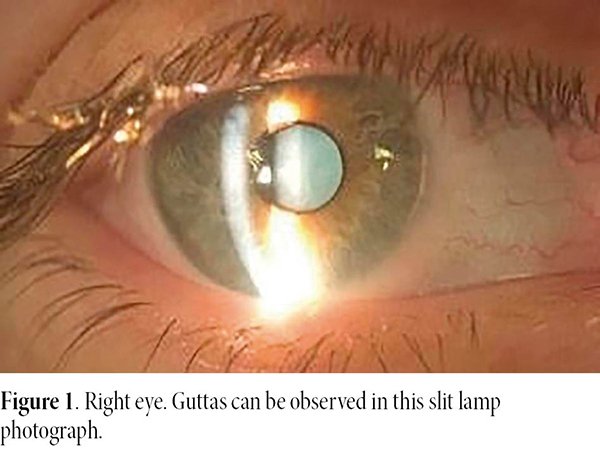
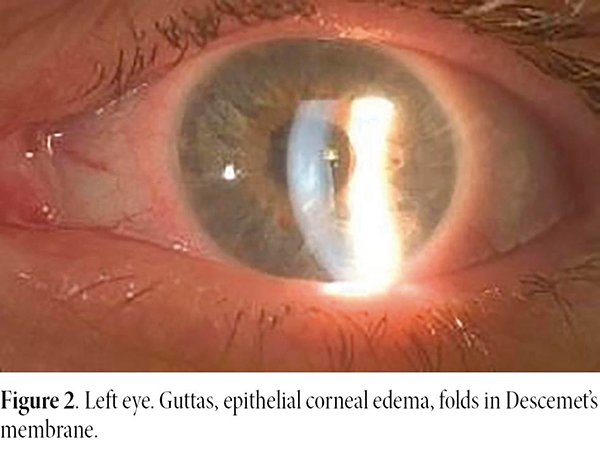
An ophthalmological evaluation was carried out in both eyes, in which the slit lamp biomicroscopy revealed central corneal guttata on the right eye and presence of central corneal guttata and clinical corneal edema and, plus folds in the Descemet’s membrane on the left eye.
At that time, specular endothelial microscopy was not performed because of the corneal edema.
The patient was referred to the corneal service. She was requested to attend with electromyogram results previously ordered by the general practitioner.
Results
As systemic findings associated with MD1, the patient presented motor impairment with walking difficulty. The electromyogram showed signs of primary muscle compromise with resting myotonic discharges. She has no hereditary family antecedents of MD1. On a physical exam, the patient presented a typical MD face with no other notable findings.
On an ophthalmologic examination, she presented best corrected visual acuity of 20/20 in the right eye and 20/40 in the left eye. Biomicroscopy revealed guttas on the right eye, and the presence of guttas, corneal edema and folds in Descemet’s membrane on the left one (Figs. 1-2). Intraocular pressure on the right eye was 12 mmHg and on the left eye 10 mmHg. The eye fundus was within normal limits in both eyes.
After starting topical treatment with 5% sodium chloride drops a good response was observed, recovering corneal transparency and absence of folds in the following two days. The dose of 5% sodium chloride drops was used every 6 hours for one month, then every 12 hours for 6 months, and then once a day every 24 hours in the morning for two years. This was the only treatment indicated both in the acute phase and in the two-year follow-up.
In the specular microscopy, carried out 7 days after the first appointment, cell density of 2807.3/mm2 was verified on the right eye, coefficient of variation 27.6%, hexagonality 56% in a total of 120 cells evaluated. Central corneal thickness was 583 microns. In the left eye: cell density of 1873.5/mm2, coefficient of variation 37.9%, hexagonality 42%, 68 cells were evaluated. Central corneal thickness was 551 microns in the left eye (Figs. 3-4). Some guttae were seen as dark spots in both eyes. Based on this, the diagnosis of FECD was confirmed in the context of a patient with a previous diagnosis of MD. Currently and after 2 years of follow-up, the patient has kept both corneas transparent, she continues using 5% sodium chloride eye drops twice daily on the left eye.


Discussion
MD is a syndrome with multisystem affection that includes myotonia, muscular dystrophy, cardiac conduction defects, endocrine and ophthalmological disorders2. MD is classified in type 1 and type 2. Type 1 MD, Steinert-Curschmann disease, Batten-Gibb disease, atrophic myotonia, or Steinert disease (MD1)3, affects chromosome 19, and type 2 MD affects chromosome 32. MD1 is the most common adult muscular dystrophy with a prevalence of 1:8000.
The medical literature describes that the most frequent ophthalmological finding is the presence of multicolored iridescent cataract with a “Christmas tree” pattern as, in general, the first manifestation of the disease. This kind of cataract is a posterior subcapsular cataract that sometimes has an early onset, without any other signs associated2. Anterior capsular contraction syndrome or early capsular phimosis can also occur in these patients after cataract surgery and intraocular lens implantation in the capsular bag4. However, until 2014, no relationship has been reported between FECD and MD. An increase in corneal thickness and a decrease in the coefficient of variability have been described in patients with MD15-6. Gattey et al. described the clinical characteristics of FECD in four patients with MD, suggesting the probability of a common pathophysiological mechanism5. Later, in a study by Winkler et al., it was observed that the prevalence of FECD was significantly higher in patients with MD1 than in the general population7. Concerning the pathogenesis, current data strongly support the premise of a shared pathophysiological mechanism of RNA toxicity between FECD and MD. Recent studies show a statistically significant correlation between both entities with proven genetic bases in which it was possible to demonstrate RNA toxicity due to the expansion of the non-coding CTG trinucleotide repeat present in both FECD and MD5,8. It is worth remembering that FECD is mostly an autosomal dominant disease, with a prevalence of approximately 4% in people in their 40s and older5,8. Clinical manifestations include loss of corneal endothelial cells, stromal edema, and a thickening of Descemet’s membrane with focal excrescences (guttas). This is due to the loss and dysfunction of corneal endothelial cells and the consequent inability to maintain dehydration of the corneal stroma and loss of corneal transparency5.
In conclusion, in patients with MD there may be an association with FECD as a cause of corneal edema. Therefore, it is essential to carry out periodic ophthalmological controls. On the other hand, considering the association of MD1 with the development of cataract, it is suggested (to perform) specular microscopy be performed before carrying out any surgical procedure in order to evaluate the corneal endothelium prior to the intervention.
References
1. Kumar A, Agarwal S, Agarwal D, Phadke SR. Myotonic dystrophy type 1 (DM1): a triplet repeat expansion disorder. Gene 2013; 522: 226-230.
2. Meola G, Cardani R. Myotonic dystrophies: an update on clinical aspects, genetic, pathology, and molecular pathomechanisms. Biochim Biophys Acta 2015; 1852: 594-606.
3. Gutiérrez Gutiérrez G, Díaz-Manera J, Almendrote M et al. Clinical guide for the diagnosis and follow-up of myotonic dystrophy type 1, MD1 or Steinert’s disease. Neurología (Engl Ed) 2020; 35: 185-206.
4. Rosa N, Lanza M, De Bernardo M et al. Anterior capsule phimosis and capsular block syndrome in a patient with Steinert myotonic dystrophy: a case report. Cases J 2009; 2: 9298.
5. Gattey D, Zhu AY, Stagner A et al. Fuchs endothelial corneal dystrophy in patients with myotonic dystrophy: a case series. Cornea 2014; 33: 96-98.
6. Rosa N, Lanza M, Borrelli M et al. Corneal thickness and endothelial cell characteristics in patients with myotonic dystrophy. Ophthalmology 2010; 117: 223-225.
7. Winkler NS, Milone M, Martinez-Thompson JM et al. Fuchs’ endothelial corneal dystrophy in patients with myotonic dystrophy, type 1. Invest Ophthalmol Vis Sci 2018; 59: 3053-3057.
8. Mootha VV, Hansen B, Rong Z et al. Fuchs’ endothelial corneal dystrophy and RNA foci in patients with myotonic dystrophy. Invest Ophthalmol Vis Sci 2017; 58: 4579-4585.