REPORTE DE CASO
Retinitis pigmentosa unilateral: reporte de caso
María Alejandra Gómez, Javier Marengo
Servicio de Oftalmología, Hospital Angel C. Padilla, San Miguel de Tucumán, Argentina.
Recibido: 16 de julio de 2019.
Aprobado: 3 de diciembre de 2019.
Correspondencia:
m.alejandragomez@hotmail.com
RESUMEN
Objetivo: Presentación de caso clínico de paciente con sospecha de retinopatía pigmentaria unilateral.
Caso clínico: Mujer de 58 años consulta por disminución de agudeza visual (AV) progresiva de 6 años de evolución. Refería haber sufrido un accidente cerebral isquémico tiempo atrás. Presentaba: AV sin corrección: ojo derecho (OD) 7/10, ojo izquierdo (OI) 2/10; con corrección: OD 10/10, OI: 5/10. Examen biomicroscópico normal ambos ojos. Fondo de ojos en OD de aspecto normal; OI: flóculos vítreos, papila ligeramente pálida, vasos con diámetro disminuidos, atenuación del brillo foveal y pigmentos en forma de osteoblastos en los cuatro cuadrantes retinales. Electrorretinograma (ERG): normal del lado derecho y mala morfología del lado izquierdo, amplitud en la respuesta retinal significativamente disminuida del lado izquierdo. Los exámenes complementarios y la serología para enfermedades infecciosas fueron negativos. Se realizó el diagnóstico diferencial con la retinosis pigmentaria típica y otras formas atípicas, así́ como con otras retinopatías pigmentarias secundarias.
Conclusión: La retinitis pigmentosa unilateral es poco frecuente y su diagnóstico se basa en el aspecto del fondo de ojos y en la alteración unilateral del ERG, excluyendo causas infecciosas, inflamatorias y vasculares. A pesar de los avances en la obtención de imágenes y las pruebas, esta patología sigue siendo un desafío diagnóstico debido a su manifestación heterogénea.
Palabras clave: retinitis pigmentosa, unilateral.
Unilateral retinitis pigmentosa: a casereport
ABSTRACT
Objective: Clinical case report of a patient with suspicion of unilateral pigmentary retinopathy.
Clinical case: 58-year-old woman presenting with progressive visual acuity (VA) loss of 6 years of evolution who referred a history of ischemic brain stroke that had occurred sometime before. Examination revealed: uncorrected VA: right eye (RE): 7/10, left eye (LE): 2/10; spectacle-corrected VA: RE: 10/10, LE: 5/10. Biomicroscopy: normal in both eyes. Funduscopy: RE: normal appearance, LE: vitreous floaters, mildly pale optic disc, decreased vessel diameter, reduced foveal brightness and osteoblast-shaped pigments in the four retinal quadrants. Electroretinogram (ERG): normal on the right side, abnormal morphology on the left side and significantly decreased retinal response amplitude on the left side. Additional tests and serology for infectious disease yielded negative results. Differential diagnosis with typical and other atypical forms of retinitis pigmentosa, as well as with other secondary pigmentary retinopathies, was made.
Conclusion: Unilateral retinitis pigmentosa is rare and its diagnosis is based on the appearance of the eye fundus and on unilateral ERG abnormalities. The presence of infections, inflammatory and vascular causes should be ruled out. In spite of advances made in image acquisition and tests, this disease is still a challenge due to its heterogeneous manifestation.
Keywords: retinitis pigmentosa, unilateral.
Retinite pigmentosa unilateral: reporte de um caso
RESUMO
Objetivo: Apresentação de caso clínico de paciente com suspeita de retinose pigmentar unilateral.
Caso clínico: Mulher de 58 anos consulta por diminuição de acuidade visual (AV) progressiva de 6 anos de evolução. Referia ter sofrido, tempo atrás, um acidente cerebral isquêmico. Apresentava: AV sem correção: olho direito (OD) 7/10, olho esquerdo (OI) 2/10; com correção: OD 10/10, OI: 5/10. Exame biomicroscópico normal ambos os olhos. Fundo de olhos em OD de aspecto normal; OI: flocos vítreos, papila ligeiramente pálida, vasos com diâmetro diminuídos, atenuação do brilho foveal e pigmentos em forma de osteoblastos nos quatro quadrantes retinianos. Electrorretinograma (ERG): normal do lado direito e má morfologia do lado esquerdo, amplitude na resposta retiniana significativamente diminuída do lado esquerdo. Os exames complementários e a serologia para doenças infeciosas foram negativos. Realizou-se o diagnóstico diferencial com a retinose pigmentar típica e outras formas atípicas, bem como com outras retinoses pigmentares secundárias.
Conclusão: A retinite pigmentosa unilateral é pouco frequente e seu diagnóstico se baseia no aspecto do fundo de olhos e na alteração unilateral do ERG, excluindo causas infeciosas, inflamatórias e vasculares. Apesar dos avanços na obtenção de imagens e nas provas, esta patologia segue sendo um desafio diagnóstico devido a sua manifestação heterogénea.
Palavras chave: retinite pigmentosa, unilateral.
INTRODUCCIÓN
La retinitis pigmentosa (RP) es un grupo de trastornos hereditarios conocidos por disfunción visual progresiva, pérdida de células y finalmente atrofia del tejido retinal. Afecta a una de cada 3.500-5.000 personas en todo el mundo1. La etiología de la RP unilateral es desconocida y se supone que es el resultado de una mutación somática durante la embriogénesis que afecta a un porcentaje de células en el cuerpo del paciente.
Dependiendo de las células involucradas, el paciente tiene la posibilidad de desarrollar esta forma atípica de RP unilateral durante su vida adulta si estas células están destinadas a convertirse en retina y RPE2. Se describe una serie de criterios para confirmar una retinitis pigmentosa monocular (RPM)3:
- Presencia en el ojo afectado de cambios funcionales y oftalmológicos de una RP típica.
- Ausencia en el otro ojo de síntomas y signos de RP.
- Período de observación mayor de 5 años que permita comprobar que el ojo contralateral no presenta la enfermedad.
- Exclusión de toda posible causa inflamatoria en el ojo afectado.
Por lo tanto, el objetivo de este trabajo es presentar un caso clínico de RP unilateral y repasar su algoritmo diagnóstico.
CASO CLÍNICO
Se recibe paciente de sexo femenino de 58 años que refiere disminución de agudeza visual progresiva de 6 años de evolución. Como antecedente de importancia mencionó haber sufrido un accidente cerebral isquémico tiempo atrás.
Al examen oftalmológico presenta: AV sin corrección OD 7/10, OI 2/10; con corrección, OD 10/10 y OI 5/10. Examen biomicroscópico normal ambos ojos. Fondo de ojos: OD: papila rosada, bordes netos, excavada 0,3, vasos de trayecto y calibre conservado, mácula con buen brillo (fig. 1); OI: vítreo celular y con flóculos, papila ligeramente pálida, vasos con diámetro disminuidos, atenuación del brillo foveal, coroides visible y pigmentos con forma de osteoblastos en los cuatro cuadrantes retinales (figs. 2 y 3). Se solicita electrorretinograma, campo visual y RFG (figs. 4 y 5). Se interroga a la paciente nuevamente y se solicita lleve a consulta exámenes previos.
Electrorretinograma informa: estudio realizado por estimulación monocular con luz blanca de alta intensidad (flash), buena morfología y reproductibilidad del lado derecho y mala morfología del lado izquierdo. La amplitud en la respuesta retinal se encontró significativamente disminuida del lado izquierdo. Los exámenes complementarios de rutina no arrojaron alteraciones y las pruebas serológicas para enfermedades infecciosas fueron negativas. Se realizó el diagnóstico diferencial con la retinosis pigmentaria típica y otras formas atípicas, así como con otras retinopatías pigmentarias secundarias
.

Figura 1. Ojo derecho.

Figura 2. Ojo izquierdo.
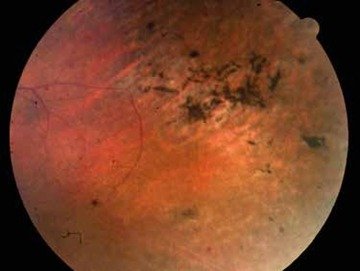
Figura 3. Periferia del ojo izquierdo.

Figura 4. Retinofluoresceinografía de ojo derecho.

Figura 5. Retinofluoresceinografía de ojo izquierdo.
DISCUSIÓN
La RP unilateral es una forma de manifestación rara de la distrofia de bastón que se describió por primera vez en 19484. Dado que las distrofias retinales suelen ser bilaterales debido a su fondo genético, una manifestación unilateral requiere de una explicación: una de ellas es la aparición de los mosaicos genéticos, es decir, la mutación afecta sólo a algunas de las células; y el segundo mecanismo es una mutación somática en lugar de una mutación de la línea germinal5. La retinosis pigmentaria unilateral es una degeneración retinal rara y esporádica causada por una mutación durante la embriogénesis. Una forma verdadera de retinosis unilateral es difícil y rara de diagnosticar, ya que, además del hecho de que en muchos casos demuestra ser una forma de degeneración de la retina debido a causas secundarias, también necesita un largo período de seguimiento para excluir una forma asimétrica bilateral de RP. Por lo tanto, una historia personal y familiar completa demuestra su utilidad en este diagnóstico diferencial2. Para poder afirmar que se está en presencia de un caso, se deben descartar todas las posibles unilateral es una degeneración retinal rara y esporádica causada por una mutación durante la embriogénesis. Una forma verdadera de retinosis unilateral es difícil y rara de diagnosticar, ya que, además del hecho de que en muchos casos demuestra ser una forma de degeneración de la retina debido a causas secundarias, también necesita un largo período de seguimiento para excluir una forma asimétrica bilateral de RP. Por lo tanto, una historia personal y familiar completa demuestra su utilidad en este diagnóstico diferencial2. Para poder afirmar que se está en presencia de un caso, se deben descartar todas las posibles etiologías infecciosas, así como tener la certeza de que los síntomas y signos se encuentran ausentes por completo en el ojo contralateral. El ERG estandarizado y el EOG son muy útiles para el diagnóstico correcto de esta afección6. La seudorretinitis pigmentaria puede provocarse por un traumatismo, coriorretinitis por sífilis, drogas como la tioridazina, cloroquina e hidrocloroquina, o por desprendimiento de retina7. Por lo tanto, para establecer el diagnóstico de retinosis pigmentaria habrá que descartar todas estas causas.
El presente caso en particular no refiere antecedentes familiares de RP, ni ciegos de causa desconocida; continúa con un OD normal mientras el OI presenta las características evolutivas típicas de la enfermedad, lo que permite afirmar que se está en presencia de una retinosis pigmentaria del OI después de un período de 5 años.
La RP unilateral es una degeneración retinal esporádica y rara causada por una mutación somática durante la embriogénesis. Una verdadera forma de RP unilateral es difícil y rara de diagnosticar ya que, además del hecho de que en muchos casos demuestra ser una forma de degeneración de la retina debido a causas secundarias, también requiere un largo período de seguimiento para excluir una forma asimétrica bilateral de RP. Por lo tanto, una historia personal y familiar completa demuestra su utilidad en este diagnóstico diferencial.
Para concluir, a pesar de los avances en la obtención de imágenes y las pruebas, las RP siguen siendo un desafío de diagnóstico debido a su heterogeneidad sustancial. La misma mutación genética puede dar lugar a diferentes manifestaciones clínicas en distintos individuos, mientras que las mismas manifestaciones se pueden deber a mutaciones dispares8.
REFERENCIAS
- Chang S, Vaccarella L, Olatunji S et al. Diagnostic challenges in retinitis pigmentosa: genotypic multiplicity and phenotypic variability. Curr Genomics 2011; 12: 267-75.
- Stamate AC, Burcea M, Zemba M. Unilateral pigmentary retinopathy: a review of literature and case presentation. Rom J Ophthalmol 2016; 60: 47-52.
- Chen H, Wu D, Huang S, Jiang F. Unilateral retinitis pigmentosa with amblyopia in the fellow eye. Graefes Arch Clin Exp Ophthalmol 2006; 244: 1701-4.
- Dreisler KK. Unilateral retinitis pigmentosa: two cases. Acta Ophthalmol 1948; 26: 385-93.
- Marsiglia M, Duncker T, Peiretti E et al. Unilateral retinitis pigmentosa: a proposal of genetic pathogenic mechanisms. Eur J Ophthalmol 2012; 22: 654-60.
- Joseph R. Unilateral retinitis pigmentosa. Br J Ophthalmol 1951; 35: 98-113.
- Sadiq MN, Bhatia J, El Batarny A, Wali UK. Unilateral retinitis pigmentosa in one eye and tilted hypoplastic disc in the other eye (two in one disease). Pak J Ophthalmol 2007; 23: 103-6.
- Nguyen H, Sujirakul T, Kulkarni N, Tsang S. Diagnosis and treatment of this genetic cause of photoreceptor degeneration. Retinal Physician 2013; 10: 34-42.